Wu Meijing, Shang Xiaobin, Sun Yue, Wu Jing, Liu Guoyan
Department of Gynecology and Obstetrics, Tianjin Medical University General Hospital, Tianjin, China.
Department of Esophageal Cancer, Tianjin Medical University Cancer Institute and Hospital, Tianjin, China.
PeerJ. 2020 May 7;8:e8961. doi: 10.7717/peerj.8961. eCollection 2020.
Abnormal expression of long non-coding RNAs (lncRNA) play a significant role in the incidence and progression of high-grade serous ovarian cancer (HGSOC), which is a leading cause of mortality among gynecologic malignant tumor patients. In this study, our aim is to identify lncRNA-associated competing endogenous RNA (ceRNA ) axes that could define more reliable prognostic parameters of HGSOC, and to investigate the lncRNAs' potential mechanism of in lymphocyte infiltration.
The RNA-seq and miRNA expression profiles were downloaded from The Cancer Genome Atlas (TCGA) and the Genotype-Tissue Expression (GTEx) database; while for obtaining the differentially expressed lncRNAs (DELs), miRNAs (DEMs), and genes (DEGs), we used edgeR, limma and DESeq2. After validating the RNA, miRNA and gene expressions, using integrated three RNA expression profiles (GSE18520, GSE27651, GSE54388) and miRNA profile (GSE47841) from the Gene Expression Omnibus (GEO) database, we performed Gene Ontology (GO) and Kyoto Encyclopedia of Gene and Genome (KEGG) pathway analyses through ClusterProfiler. The prognostic value of these genes was determined with Kaplan-Meier survival analysis and Cox regression analysis. The ceRNA network was constructed using Cytoscape. The correlation between lncRNAs in ceRNA network and immune infiltrating cells was analyzed by using Tumor IMmune Estimation Resource (TIMER), and gene markers of tumor-infiltrating immune cells were identified using Spearman's correlation after removing the influence of tumor purity.
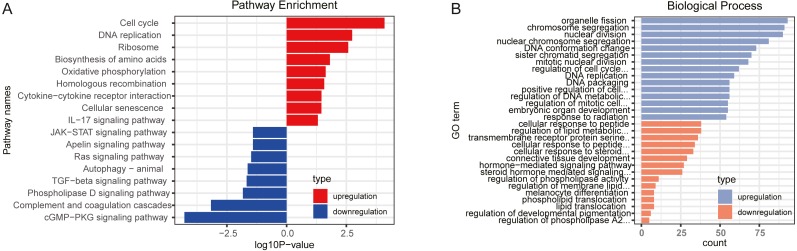
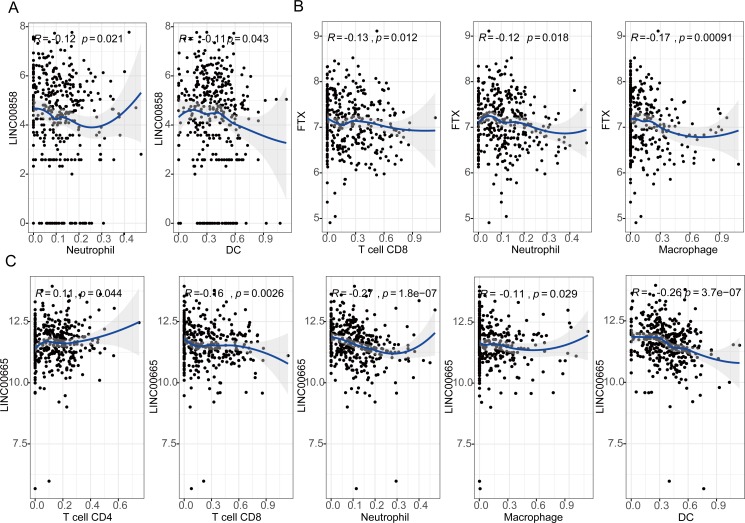
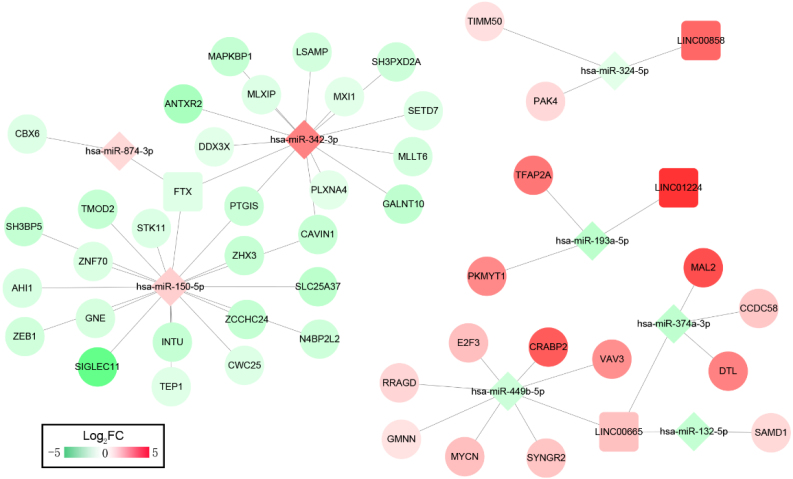
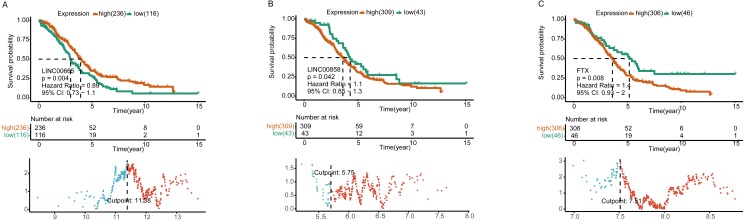
A total of 33 DELs (25 upregulated and eight downregulated), 134 DEMs (76 upregulated and 58 downregulated), and 1,612 DEGs (949 upregulated and 663 downregulated) were detected that could be positively correlated with overall survival (OS) of HGSOC. With the 1,612 analyzed genes, we constructed a ceRNA network, which indicated a pre-dominant involvement of the immune-related pathways. Furthermore, our data revealed that LINC00665 influenced the infiltration level of macrophages and dendritic cells (DCs). On the other hand, FTX and LINC00665, which may play their possible roles through the ceRNA axis, demonstrated a potential to inhibit Tregs and prevent T-cell exhaustion.
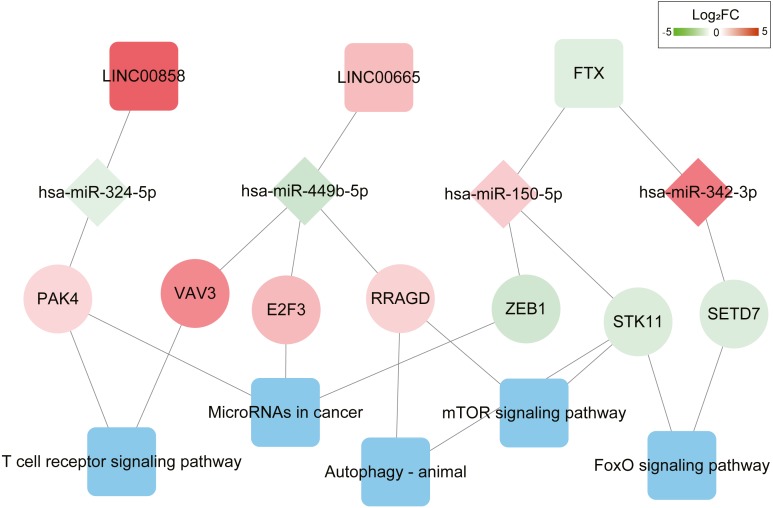
We defined several prognostic biomarkers for the incidence and progression of HGSOC and constructed a network for ceRNA axes; among which three were indicated to have a positive correlation with lymphocyte infiltration, namely: FTX-hsa-miR-150-5p-STK11, LINC00665-hsa-miR449b-5p-VAV3 and LINC00665-hsa-miR449b-5p-RRAGD.
长链非编码RNA(lncRNA)的异常表达在高级别浆液性卵巢癌(HGSOC)的发生和发展中起重要作用,HGSOC是妇科恶性肿瘤患者死亡的主要原因。在本研究中,我们的目的是鉴定lncRNA相关的竞争性内源性RNA(ceRNA)轴,其可定义HGSOC更可靠的预后参数,并研究lncRNA在淋巴细胞浸润中的潜在机制。
从癌症基因组图谱(TCGA)和基因型-组织表达(GTEx)数据库下载RNA测序和miRNA表达谱;为了获得差异表达的lncRNA(DEL)、miRNA(DEM)和基因(DEG),我们使用了edgeR、limma和DESeq2。在验证RNA、miRNA和基因表达后,使用来自基因表达综合数据库(GEO)的三个RNA表达谱(GSE18520、GSE27651、GSE54388)和miRNA谱(GSE47841),我们通过ClusterProfiler进行基因本体(GO)和京都基因与基因组百科全书(KEGG)通路分析。通过Kaplan-Meier生存分析和Cox回归分析确定这些基因的预后价值。使用Cytoscape构建ceRNA网络。使用肿瘤免疫估计资源(TIMER)分析ceRNA网络中lncRNA与免疫浸润细胞之间的相关性,并在消除肿瘤纯度影响后使用Spearman相关性鉴定肿瘤浸润免疫细胞的基因标志物。
共检测到33个DEL(25个上调和8个下调)、134个DEM(76个上调和58个下调)和1612个DEG(949个上调和663个下调),它们与HGSOC的总生存期(OS)呈正相关。利用这1612个分析基因,我们构建了一个ceRNA网络,表明其主要参与免疫相关通路。此外,我们的数据显示LINC00665影响巨噬细胞和树突状细胞(DC)的浸润水平。另一方面,FTX和LINC00665可能通过ceRNA轴发挥其可能的作用,显示出抑制调节性T细胞(Tregs)和防止T细胞耗竭的潜力。
我们定义了几个HGSOC发生和发展的预后生物标志物,并构建了ceRNA轴网络;其中三个与淋巴细胞浸润呈正相关,即:FTX-hsa-miR-150-5p-STK11、LINC00665-hsa-miR449b-5p-VAV3和LINC00665-hsa-miR449b-5p-RRAGD。