MTA-DE Laboratory of Protein Dynamics, Department of Biochemistry and Molecular Biology, University of Debrecen, Debrecen, Hungary.
The John Curtin School of Medical Research, The Australian National University, Canberra, Australia.
PLoS Comput Biol. 2020 May 26;16(5):e1007864. doi: 10.1371/journal.pcbi.1007864. eCollection 2020 May.
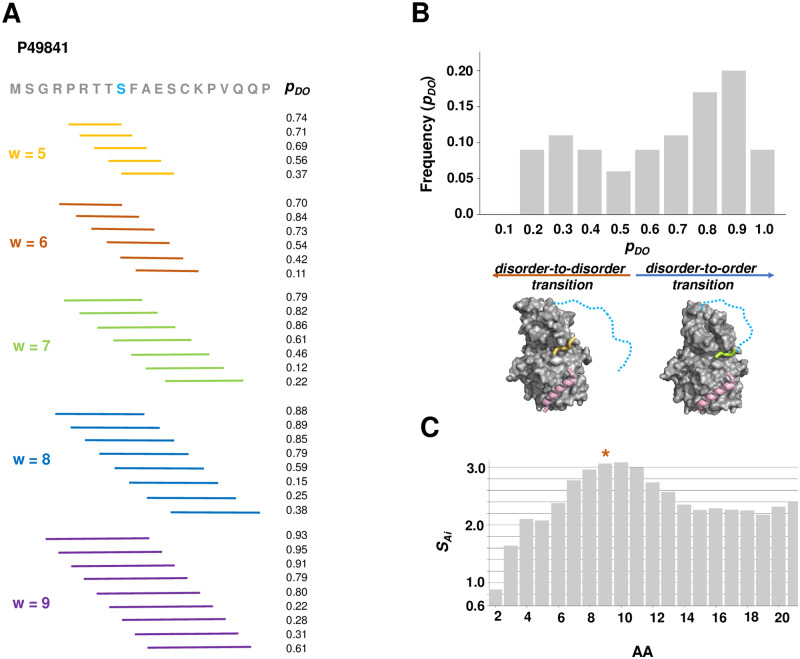
Interactions between disordered proteins involve a wide range of changes in the structure and dynamics of the partners involved. These changes can be classified in terms of binding modes, which include disorder-to-order (DO) transitions, when proteins fold upon binding, as well as disorder-to-disorder (DD) transitions, when the conformational heterogeneity is maintained in the bound states. Furthermore, systematic studies of these interactions are revealing that proteins may exhibit different binding modes with different partners. Proteins that exhibit this context-dependent binding can be referred to as fuzzy proteins. Here we investigate amino acid code for fuzzy binding in terms of the entropy of the probability distribution of transitions towards decreasing order. We implement these entropy calculations into the FuzPred (http://protdyn-fuzpred.org) algorithm to predict the range of context-dependent binding modes of proteins from their amino acid sequences. As we illustrate through a variety of examples, this method identifies those binding sites that are sensitive to the cellular context or post-translational modifications, and may serve as regulatory points of cellular pathways.
蛋白质的无序相互作用涉及到参与的伴侣结构和动力学的广泛变化。这些变化可以根据结合模式进行分类,包括无序到有序(DO)转变,即蛋白质在结合时折叠,以及无序到无序(DD)转变,即构象异质性在结合态中得以维持。此外,对这些相互作用的系统研究表明,蛋白质可能与不同的伴侣表现出不同的结合模式。表现出这种上下文相关结合的蛋白质可以被称为模糊蛋白质。在这里,我们根据向低序转变的概率分布的熵来研究模糊结合的氨基酸编码。我们将这些熵计算纳入到 FuzPred(http://protdyn-fuzpred.org)算法中,以从蛋白质的氨基酸序列预测蛋白质上下文相关结合模式的范围。正如我们通过各种例子所说明的,这种方法可以识别那些对细胞环境或翻译后修饰敏感的结合位点,并可能成为细胞途径的调节点。