Department of Pharmacology and Toxicology School of Medicine University of Mississippi Medical Center Jackson MS.
J Am Heart Assoc. 2020 Jun 2;9(11):e015895. doi: 10.1161/JAHA.120.015895. Epub 2020 May 29.
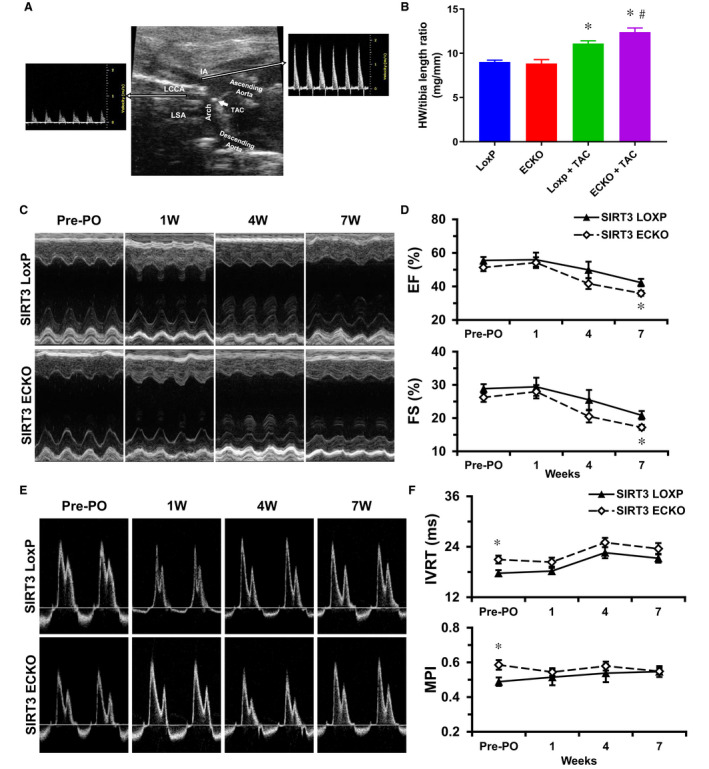
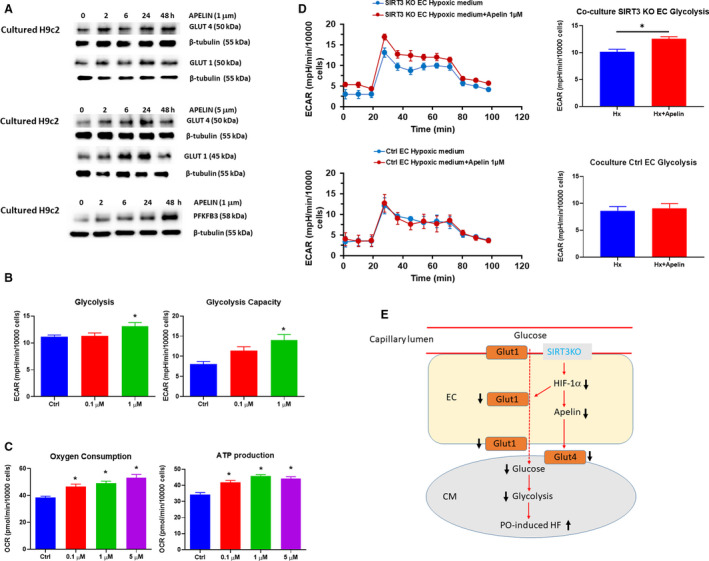
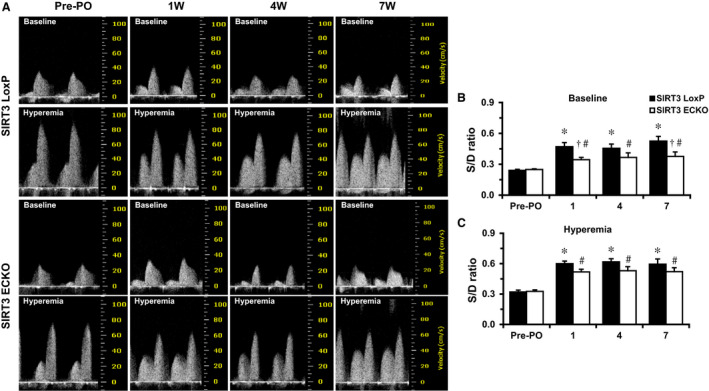
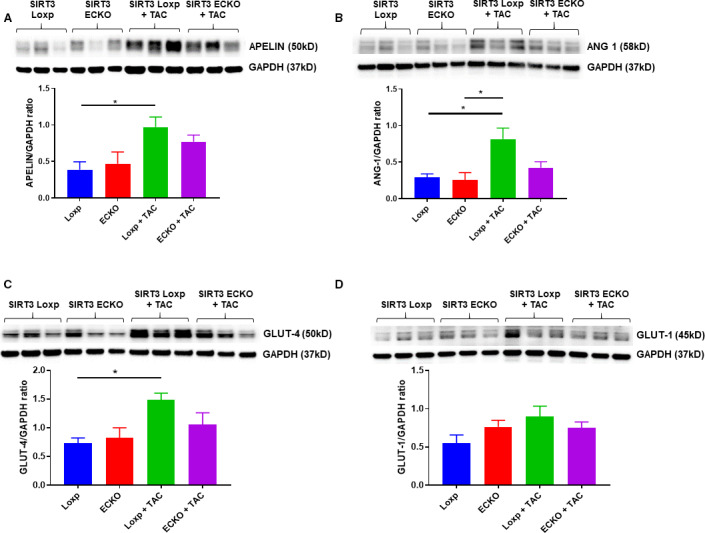
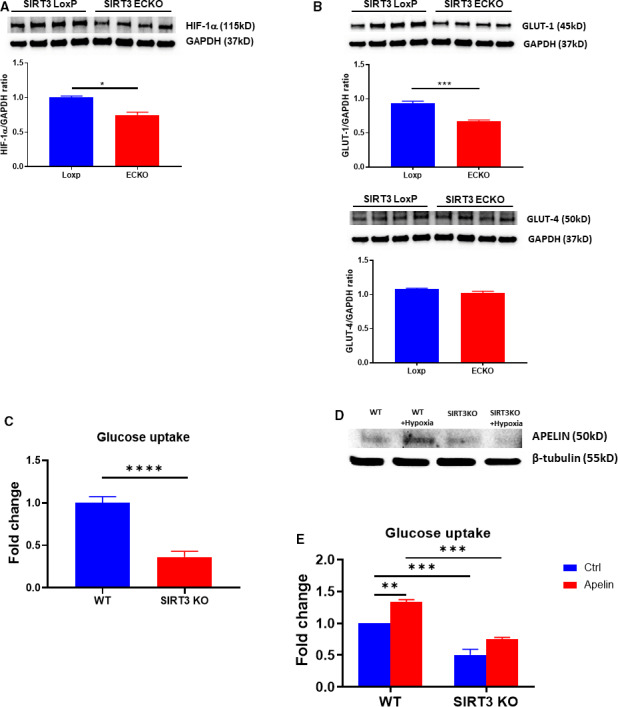
Background Alterations of energetic metabolism are suggested to be an important contributor to pressure overload (PO)-induced heart failure. Our previous study reveals that knockout of endothelial Sirtuin 3 (SIRT3) alters glycolysis and impairs diastolic function. We hypothesize that endothelial SIRT3 regulates glucose utilization of cardiomyocytes and sensitizes PO-induced heart failure. Methods and Results SIRT3 endothelial cell knockout mice and their control SIRT3 LoxP mice were subjected to PO by transverse aortic constriction for 7 weeks. The ratio of heart weight to tibia length was increased in both strains of mice, in which SIRT3 endothelial cell knockout mice+transverse aortic constriction exhibited more severe cardiac hypertrophy. Coronary blood flow and systolic function were significantly decreased in SIRT3 endothelial cell knockout mice+transverse aortic constriction compared with SIRT3 LoxP mice+transverse aortic constriction, as evidenced by lower systolic/diastolic ratio, ejection fraction, and fractional shortening. PO-induced upregulation of apelin and glucose transporter 4 were significantly reduced in the hearts of SIRT3 endothelial cell knockout mice. In vitro levels of hypoxia-inducible factor-1α and glucose transporter 1 and glucose uptake were significantly reduced in SIRT3 knockout endothelial cells. Furthermore, hypoxia-induced apelin expression was abolished together with reduced apelin-mediated glucose uptake in SIRT3 knockout endothelial cells. Exposure of cardiomyocyte with apelin increased expression of glucose transporter 1 and glucose transporter 4. This was accompanied by a significant increase in glycolysis. Supplement of apelin in SIRT3 knockout hypoxic endothelial cell media increased glycolysis in the cardiomyocytes. Conclusions Knockout of SIRT3 disrupts glucose transport from endothelial cells to cardiomyocytes, reduces cardiomyocyte glucose utilization via apelin in a paracrine manner, and sensitizes PO-induced heart failure. Endothelial SIRT3 may regulate cardiomyocyte glucose availability and govern the function of the heart.
能量代谢的改变被认为是导致压力超负荷(PO)诱导心力衰竭的一个重要因素。我们之前的研究表明,内皮细胞沉默信息调节因子 3(SIRT3)的缺失会改变糖酵解并损害舒张功能。我们假设内皮细胞 SIRT3 调节心肌细胞的葡萄糖利用,并使 PO 诱导的心力衰竭敏感化。
SIRT3 内皮细胞敲除小鼠及其对照 SIRT3 LoxP 小鼠接受了 7 周的主动脉缩窄术(transverse aortic constriction)来进行 PO 诱导。两种小鼠的心脏重量与胫骨长度的比值均增加,其中 SIRT3 内皮细胞敲除小鼠+主动脉缩窄组表现出更严重的心脏肥大。与 SIRT3 LoxP 小鼠+主动脉缩窄组相比,SIRT3 内皮细胞敲除小鼠+主动脉缩窄组的冠状动脉血流和收缩功能显著降低,表现为收缩/舒张比、射血分数和短轴缩短率降低。PO 诱导的 SIRT3 内皮细胞敲除小鼠心脏中 apelin 和葡萄糖转运蛋白 4 的上调明显减少。SIRT3 敲除内皮细胞中缺氧诱导因子-1α和葡萄糖转运蛋白 1 和葡萄糖摄取的水平显著降低。此外,SIRT3 敲除内皮细胞中的缺氧诱导的 apelin 表达被废除,同时伴随着 apelin 介导的葡萄糖摄取减少。apelin 暴露于心肌细胞中增加了葡萄糖转运蛋白 1 和葡萄糖转运蛋白 4 的表达。这伴随着糖酵解的显著增加。在 SIRT3 敲除缺氧内皮细胞培养基中补充 apelin 增加了心肌细胞中的糖酵解。
SIRT3 的缺失破坏了内皮细胞向心肌细胞的葡萄糖转运,通过旁分泌方式减少了心肌细胞的葡萄糖利用,使 PO 诱导的心力衰竭敏感化。内皮细胞 SIRT3 可能调节心肌细胞葡萄糖的可用性,并控制心脏的功能。