Department of Applied Biology, School of Applied Natural Science, Adama Science and Technology University, P.O.Box 1888, Adama, Ethiopia.
Department of Applied Biology, School of Applied Natural Science, Adama Science and Technology University, P.O.Box 1888, Adama, Ethiopia.
Infect Genet Evol. 2020 Oct;84:104386. doi: 10.1016/j.meegid.2020.104386. Epub 2020 May 29.
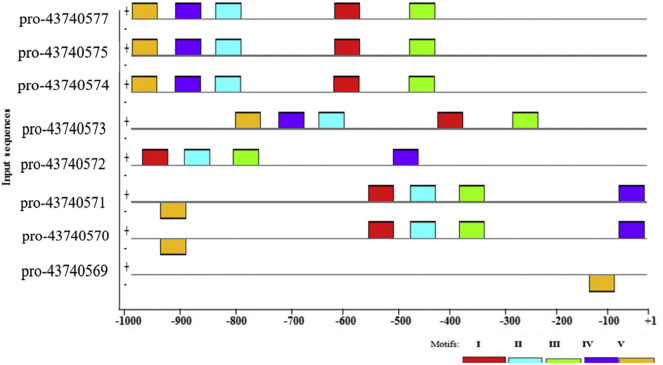
SARS-CoV-2 is a new virus responsible for an outbreak of respiratory illness known as COVID-19, which has spread to several countries around the world and a global effort is being undertaken to characterize the molecular features and evolutionary origins of this virus. In silico analysis of the transcription start sites, promoter regions, transcription factors and their binding sites, gene ontology, CpG islands for SARS-CoV-2 viral genome are a first step to understand the regulation mechanisms of gene expression and its association with genetic variations in the genomes. For this purpose, we first computationally surveyed all SARS-CoV-2 virus genes with the open reading frames from NCBI database and found eleven sequences to accomplish the mentioned features by using bioinformatics tools. Our analysis revealed that all (100%) of the SARS-CoV-2 virus genes have more than one TSS. By taking all TSSs with the highest predictive score we determined promoter regions and identified five common candidate motifs (MVI, MVII, MVIII, MVIV and MVV) of which MVI was found to be shared by all promoter regions of SARS-CoV-2 virus genes with the least E-value (3.8e-056, statistically highly significant). In our further analysis of MVI we showed MVI serve as binding sites for a single transcription factor (TF) family, EXPREG, involved in the regulatory mode of these genes. From EXPREG family four TFs that belongs to Cyclic AMP (cAMP) receptor protein (CRP) and Catabolite control protein A (CcpA) group mostly serve as transcriptional activator whereas two TFs that belong to LexA group always serve as transcriptional repressor in different kinds of cellular processes and molecular functions. Therefore, we unfolded SARS-CoV-2 viral genome to shed light on its gene expression regulation that could help to design and evaluate diagnostic tests, to track and trace the ongoing outbreak and to identify potential intervention options.
严重急性呼吸系统综合征冠状病毒 2 是一种新病毒,可引起被称为 COVID-19 的呼吸道疾病爆发,该病毒已传播到世界上多个国家,目前正在进行全球努力,以确定该病毒的分子特征和进化起源。对 SARS-CoV-2 病毒基因组的转录起始位点、启动子区域、转录因子及其结合位点、基因本体论和 CpG 岛进行计算机分析,是了解基因表达调控机制及其与基因组中遗传变异关联的第一步。为此,我们首先使用生物信息学工具,从 NCBI 数据库中计算调查了所有 SARS-CoV-2 病毒基因的开放阅读框,并找到了十一个序列来完成上述特征。我们的分析表明,所有(100%)SARS-CoV-2 病毒基因都有一个以上的 TSS。通过采用具有最高预测得分的所有 TSS,我们确定了启动子区域,并鉴定了五个常见的候选基序(MVI、MVII、MVIII、MVIV 和 MVV),其中 MVI 被发现是 SARS-CoV-2 病毒基因所有启动子区域的共有基序,其 E 值最低(3.8e-056,统计学上具有高度显著性)。在我们对 MVI 的进一步分析中,我们表明 MVI 作为单个转录因子(TF)家族 EXPREG 的结合位点,该家族参与这些基因的调控模式。在 EXPREG 家族中,属于环腺苷酸(cAMP)受体蛋白(CRP)和分解代谢物控制蛋白 A(CcpA)组的四个 TF 主要作为转录激活因子,而属于 LexA 组的两个 TF 总是作为不同细胞过程和分子功能的转录抑制因子。因此,我们对 SARS-CoV-2 病毒基因组进行了展开,以阐明其基因表达调控机制,这有助于设计和评估诊断测试、跟踪和追踪正在进行的疫情爆发,并确定潜在的干预选择。