Ponnapalli Sri Priya, Bradley Matthew W, Devine Karen, Bowen Jay, Coppens Sara E, Leraas Kristen M, Milash Brett A, Li Fuqiang, Luo Huijuan, Qiu Shi, Wu Kui, Yang Huanming, Wittwer Carl T, Palmer Cheryl A, Jensen Randy L, Gastier-Foster Julie M, Hanson Heidi A, Barnholtz-Sloan Jill S, Alter Orly
Scientific Computing and Imaging Institute, University of Utah, Salt Lake City, Utah 84112, USA.
Department of Population and Quantitative Health Sciences, Case Western Reserve University School of Medicine, Cleveland, Ohio 44106, USA.
APL Bioeng. 2020 May 15;4(2):026106. doi: 10.1063/1.5142559. eCollection 2020 Jun.
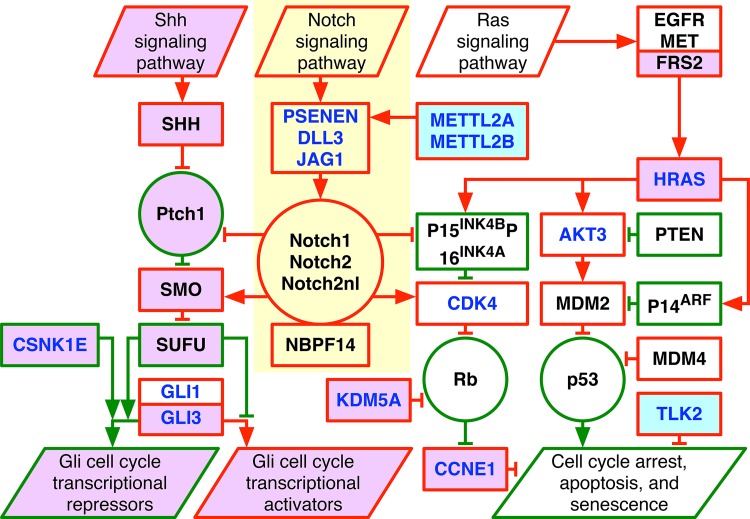
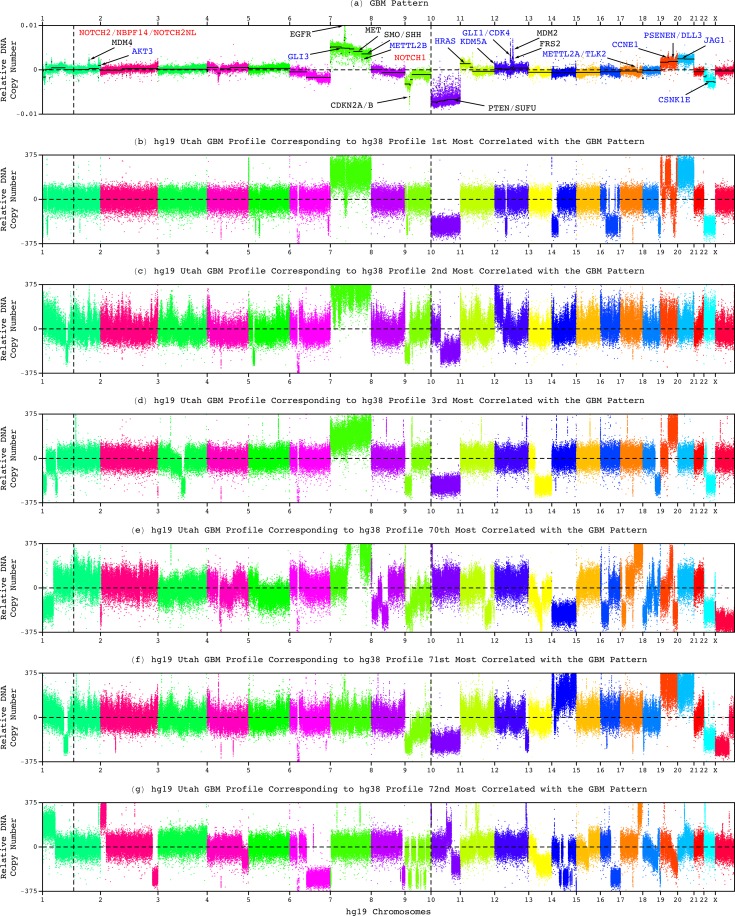
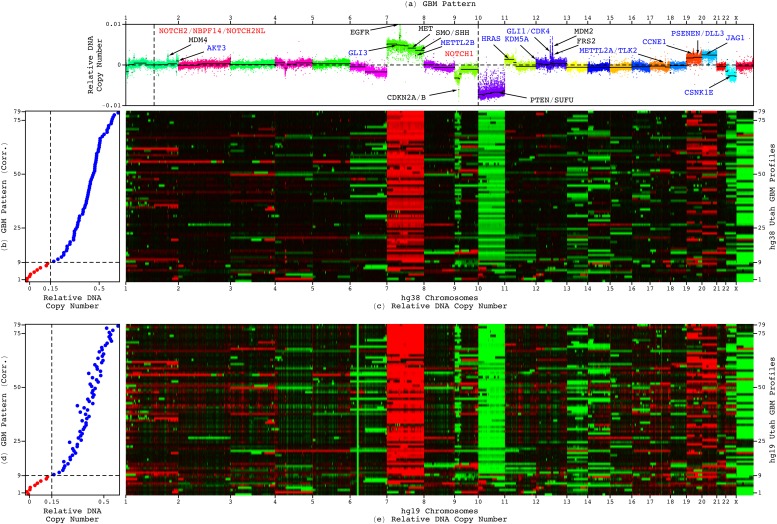
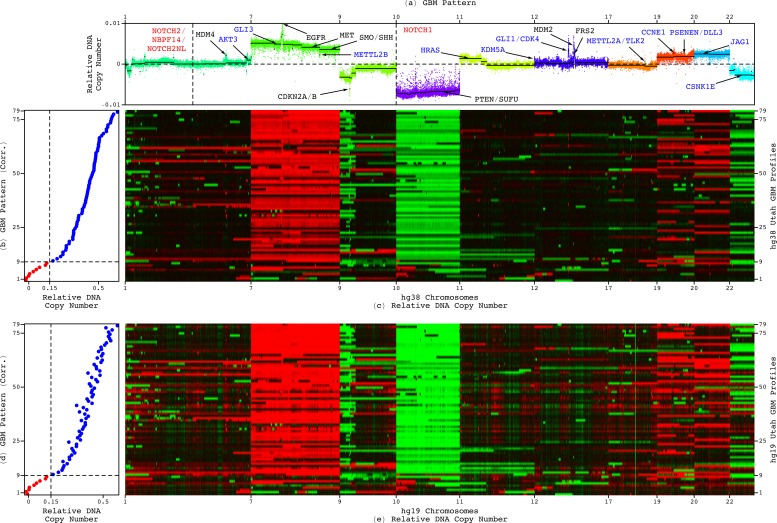
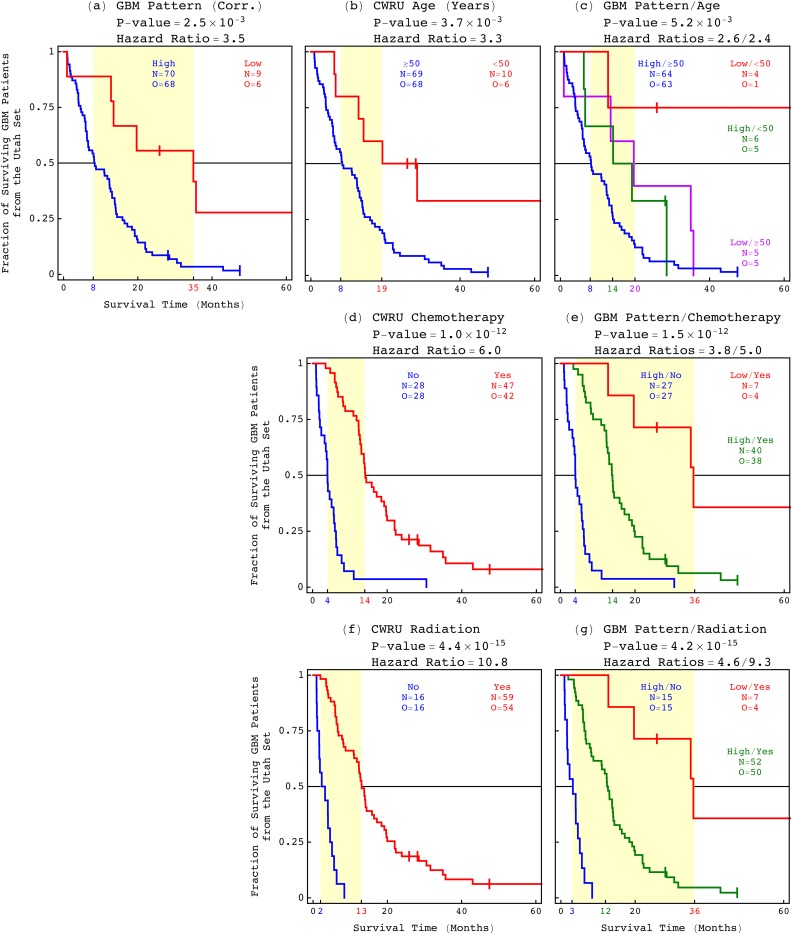
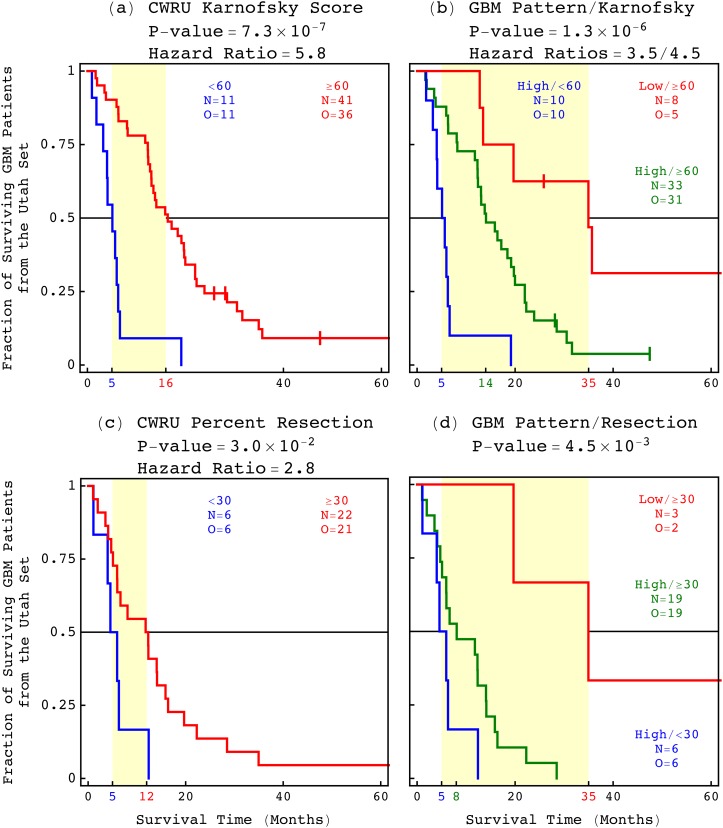
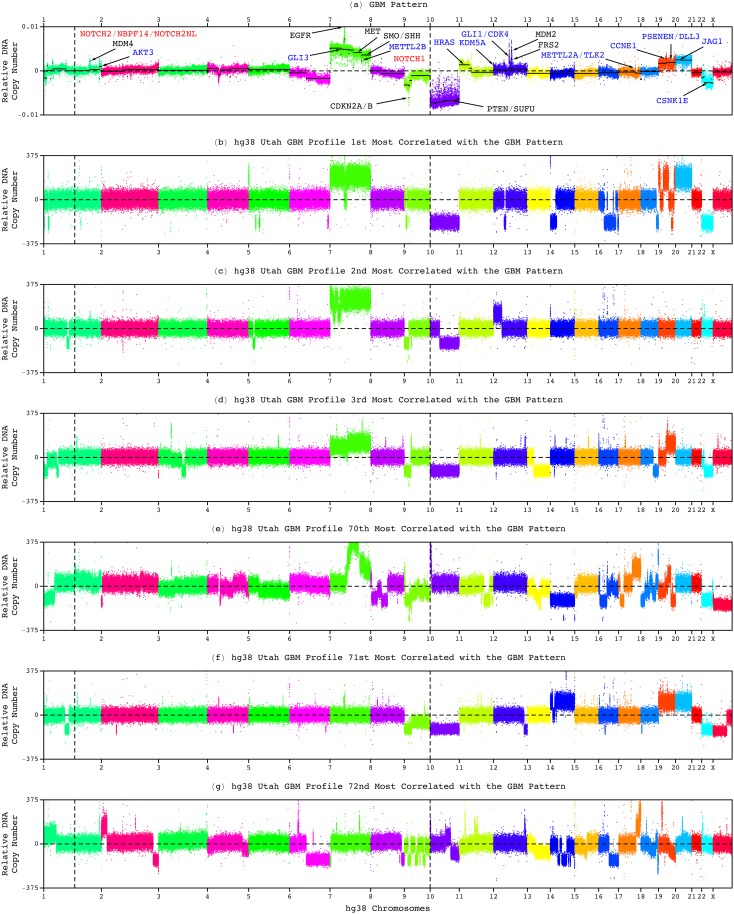
Modeling of genomic profiles from the Cancer Genome Atlas (TCGA) by using recently developed mathematical frameworks has associated a genome-wide pattern of DNA copy-number alterations with a shorter, roughly one-year, median survival time in glioblastoma (GBM) patients. Here, to experimentally test this relationship, we whole-genome sequenced DNA from tumor samples of patients. We show that the patients represent the U.S. adult GBM population in terms of most normal and disease phenotypes. Intratumor heterogeneity affects and profiling technology and reference human genome specifics affect <1% of the classifications of the tumors by the pattern, where experimental batch effects normally reduce the reproducibility, i.e., precision, of classifications based upon between one to a few hundred genomic loci by >30%. With a 2.25-year Kaplan-Meier median survival difference, a 3.5 univariate Cox hazard ratio, and a 0.78 concordance index, i.e., accuracy, the pattern predicts survival better than and independent of age at diagnosis, which has been the best indicator since 1950. The prognostic classification by the pattern may, therefore, help to manage GBM pseudoprogression. The diagnostic classification may help drugs progress to regulatory approval. The therapeutic predictions, of previously unrecognized targets that are correlated with survival, may lead to new drugs. Other methods missed this relationship in the roughly 3B-nucleotide genomes of the small, order of magnitude of 100, patient cohorts, e.g., from TCGA. Previous attempts to associate GBM genotypes with patient phenotypes were unsuccessful. This is a proof of principle that the frameworks are uniquely suitable for discovering clinically actionable genotype-phenotype relationships.
通过使用最近开发的数学框架对癌症基因组图谱(TCGA)中的基因组概况进行建模,发现胶质母细胞瘤(GBM)患者全基因组范围内的DNA拷贝数改变模式与较短的中位生存时间相关,中位生存时间约为一年。在此,为了通过实验验证这种关系,我们对患者肿瘤样本的DNA进行了全基因组测序。我们表明,就大多数正常和疾病表型而言,这些患者代表了美国成年GBM人群。肿瘤内异质性影响以及分析技术和参考人类基因组特性对基于该模式的肿瘤分类影响不到1%,而实验批次效应通常会使基于一到几百个基因组位点的分类的可重复性(即精度)降低30%以上。该模式的Kaplan-Meier中位生存差异为2.25年,单变量Cox风险比为3.5,一致性指数(即准确性)为0.78,其预测生存情况优于诊断时的年龄且与之无关,而诊断时的年龄自1950年以来一直是最佳指标。因此,该模式的预后分类可能有助于管理GBM假性进展。诊断分类可能有助于药物获得监管批准。对与生存相关的先前未被识别的靶点的治疗预测可能会催生新药。在来自TCGA等规模约为100的小患者队列的大约30亿个核苷酸的基因组中,其他方法未能发现这种关系。先前将GBM基因型与患者表型相关联的尝试均未成功。这证明了这些框架特别适合发现具有临床可操作性的基因型-表型关系。