Zhao Sheng, Zhang Cuicui, Mu Jianqiang, Zhang Hui, Yao Wen, Ding Xinhua, Ding Junqiang, Chang Yuxiao
Shenzhen Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Genome Analysis Laboratory of the Ministry of Agriculture, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen, 518120 China.
College of Life Science and Technology, Guangxi University, Nanning, 530004 China.
Plant Methods. 2020 May 24;16:74. doi: 10.1186/s13007-020-00615-3. eCollection 2020.
Next generation sequencing (NGS) has been widely used in biological research, due to its rapid decrease in cost and increasing ability to generate data. However, while the sequence generation step has seen many improvements over time, the library preparation step has not, resulting in low-efficiency library preparation methods, especially for the most time-consuming and labor-intensive steps: size-selection and quantification. Consequently, there can be bottlenecks in projects with large sample cohorts.
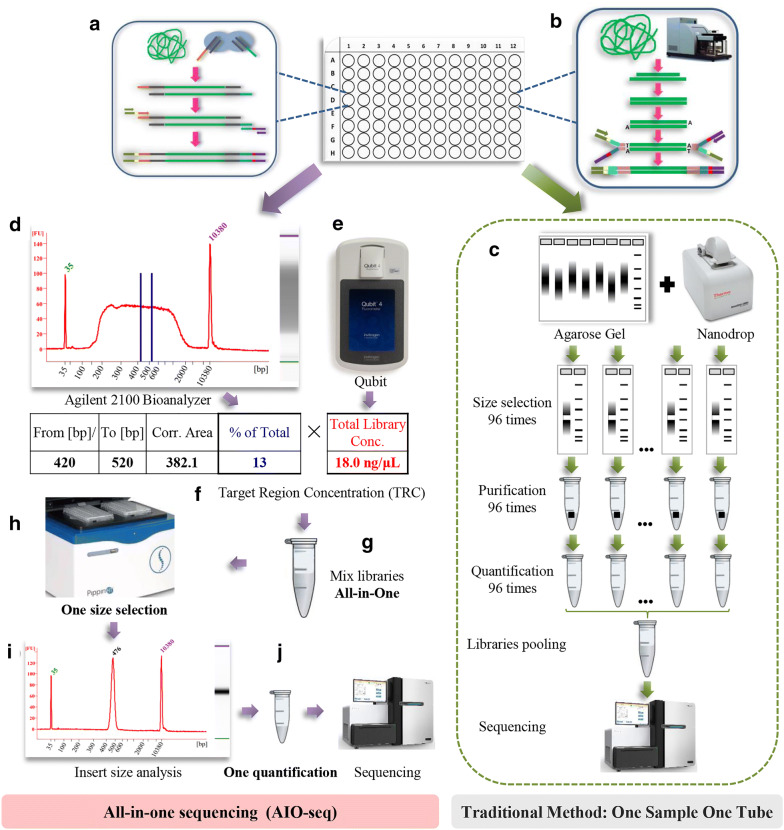
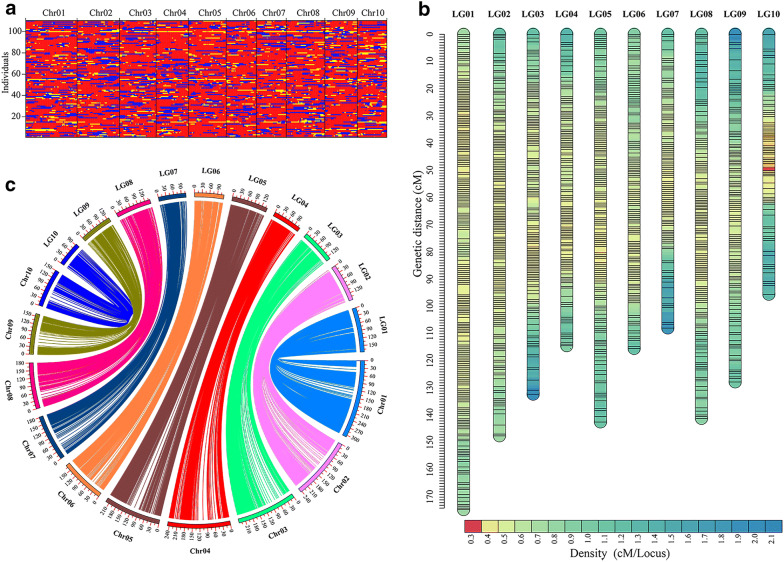
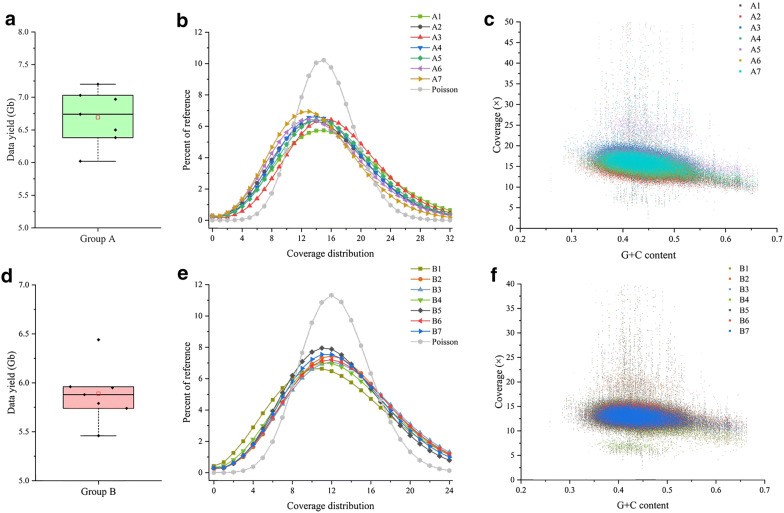
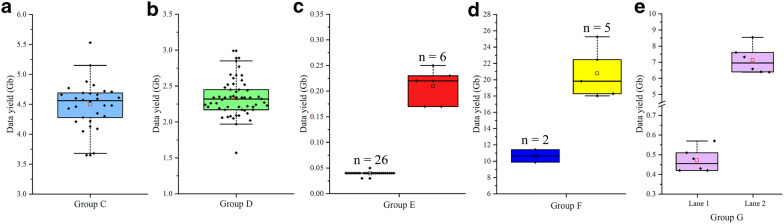
We have described the all-in-one sequencing (AIO-seq) method, where instead of performing size-selection and quantification for samples individually, one sample one tube, up to 116 samples are pooled and analyzed in a single tube, 'All-In-One'. The AIO-seq method pools libraries based on the samples' expected data yields and the calculated concentrations of the size selected regions (target region), which can easily be obtained with the Agilent 2100 Bioanalyzer and Qubit Fluorometer. AIO-seq was applied to whole genome sequencing and RNA-seq libraries successfully, and it is envisaged that it could be applied to any type of NGS library, such as chromatin immunoprecipitation coupled with massively parallel sequencing, assays for transposase-accessible chromatin with high-throughput sequencing, and high-throughput chromosome conformation capture. We also demonstrated that for genetic population samples with low coverage sequences, like recombinant inbred lines (RIL), AIO-seq could be further simplified, by mixing the libraries immediately after PCR, without calculating the target region concentrations.
The AIO-seq method is thus labor saving and cost effective, and suitable for projects with large sample cohorts, like those used in plant breeding or population genetics research.
由于成本迅速下降且生成数据的能力不断提高,新一代测序(NGS)已广泛应用于生物学研究。然而,尽管随着时间的推移序列生成步骤有了许多改进,但文库制备步骤却没有,导致文库制备方法效率低下,尤其是对于最耗时且劳动强度大的步骤:大小选择和定量。因此,在有大量样本队列的项目中可能会出现瓶颈。
我们描述了一体化测序(AIO-seq)方法,即不是对样本逐个进行大小选择和定量(一个样本一管),而是将多达116个样本合并在一个管中进行“一体化”分析。AIO-seq方法根据样本的预期数据产量和所选大小区域(目标区域)的计算浓度来合并文库,使用安捷伦2100生物分析仪和Qubit荧光计可以轻松获得这些数据。AIO-seq已成功应用于全基因组测序和RNA-seq文库,并且可以设想它可应用于任何类型的NGS文库,例如染色质免疫沉淀结合大规模平行测序、转座酶可及染色质高通量测序分析以及高通量染色体构象捕获。我们还证明,对于低覆盖度序列的遗传群体样本,如重组自交系(RIL),在PCR后立即混合文库,无需计算目标区域浓度,AIO-seq可以进一步简化。
因此,AIO-seq方法节省人力且具有成本效益,适用于有大量样本队列的项目,如植物育种或群体遗传学研究中使用的项目。