Coy Samantha R, Gann Eric R, Papoulis Spiridon E, Holder Michael E, Ajami Nadim J, Petrosino Joseph F, Zinser Erik R, Van Etten James L, Wilhelm Steven W
Department of Microbiology, The University of Tennessee, Knoxville, Knoxville, TN, United States.
BioSciences at Rice, Rice University, Houston, TX, United States.
Front Microbiol. 2020 May 19;11:887. doi: 10.3389/fmicb.2020.00887. eCollection 2020.
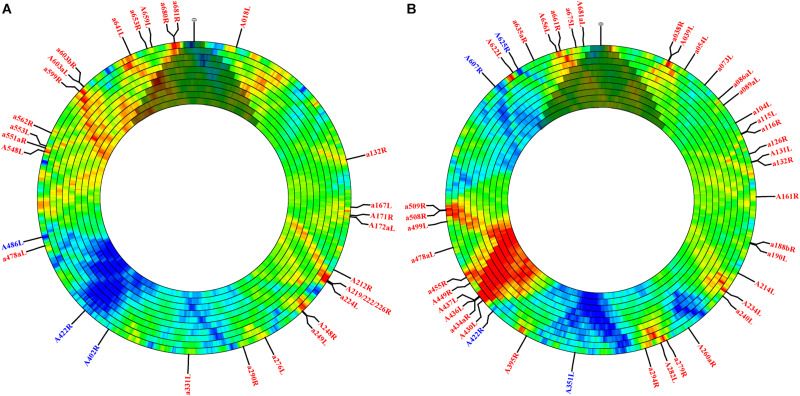
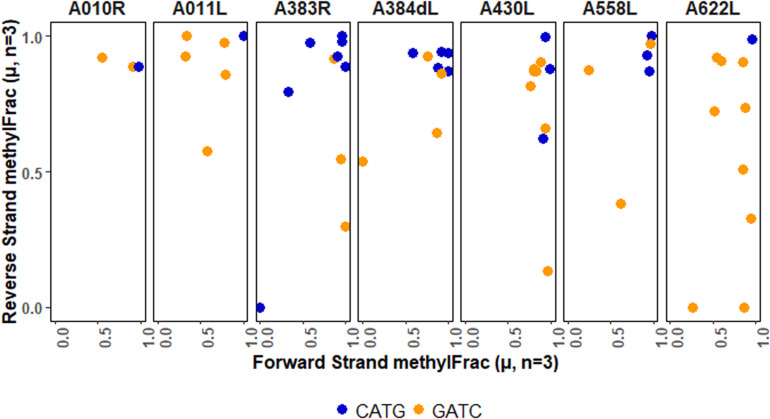
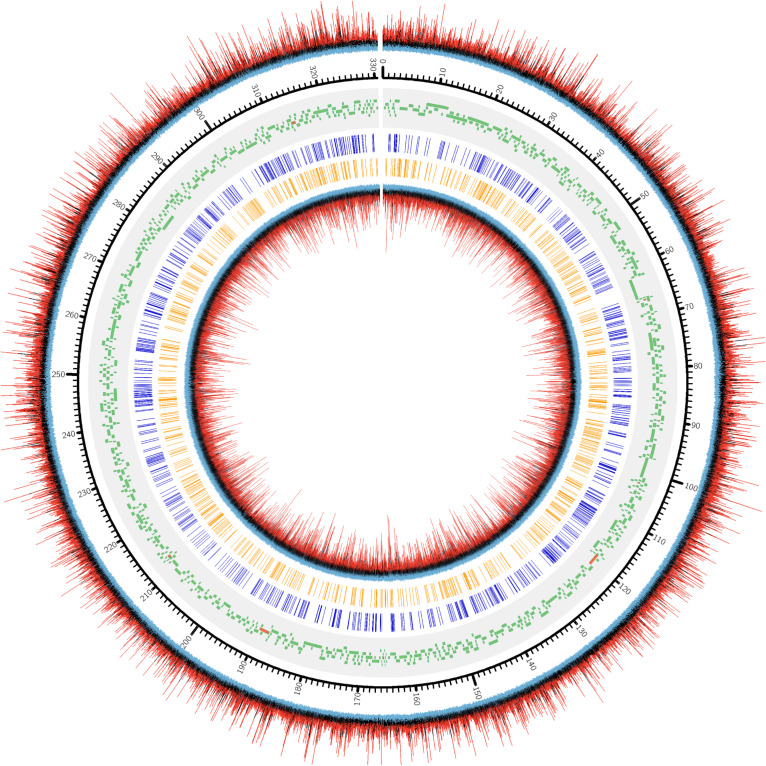
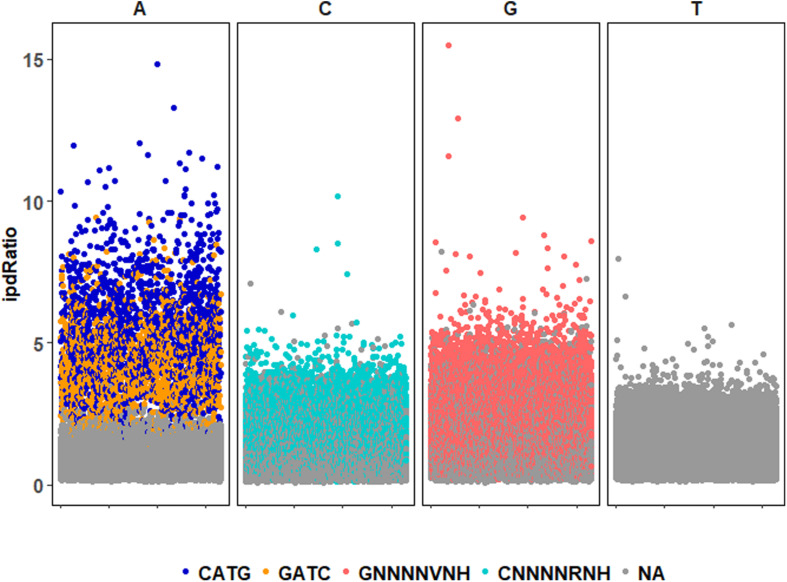
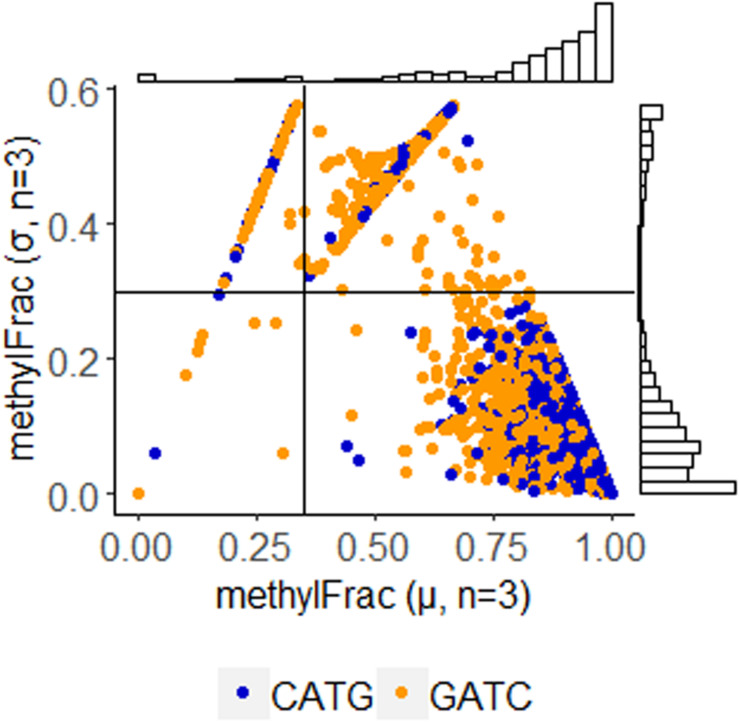
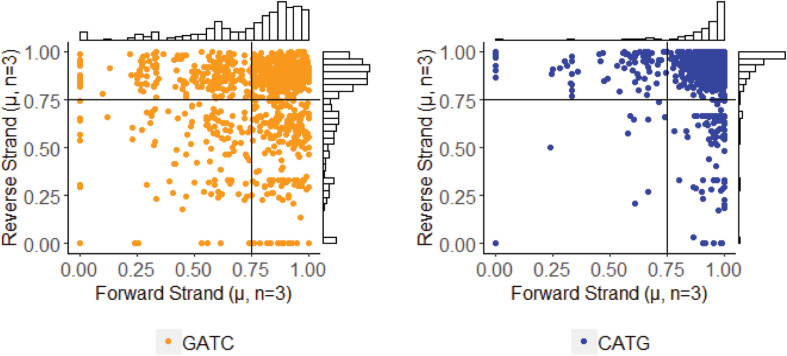
Chloroviruses (family ) infect eukaryotic, freshwater, unicellular green algae. A unique feature of these viruses is an abundance of DNA methyltransferases, with isolates dedicating up to 4.5% of their protein coding potential to these genes. This diversity highlights just one of the long-standing values of the chlorovirus model system; where group-wide epigenomic characterization might begin to elucidate the function(s) of DNA methylation in large dsDNA viruses. We characterized DNA modifications in the prototype chlorovirus, PBCV-1, using single-molecule real time (SMRT) sequencing ( PacBio). Results were compared to total available sites predicted based on DNA sequence alone. SMRT-software detected N6-methyl-adenine (m6A) at GATC and CATG recognition sites, motifs previously shown to be targeted by PBCV-1 DNA methyltransferases M.CviAI and M. AII, respectively. At the same time, PacBio analyses indicated that 10.9% of the PBCV-1 genome had large interpulse duration ratio (ipdRatio) values, the primary metric for DNA modification identification. These events represent 20.6x more sites than can be accounted for by all available adenines in GATC and CATG motifs, suggesting base or backbone modifications other than methylation might be present. To define methylation stability, we cross-compared methylation status of each GATC and CATG sequence in three biological replicates and found ∼81% of sites were stably methylated, while ∼2% consistently lack methylation. The remaining 17% of sites were stochastically methylated. When methylation status was analyzed for both strands of each target, we show that palindromes existed in completely non-methylated states, fully-methylated states, or hemi-methylated states, though GATC sites more often lack methylation than CATG sequences. Given that both sequences are targeted by not just methyltransferases, but by restriction endonucleases that are together encoded by PBCV-1 as virus-originating restriction modification (RM) systems, there is strong selective pressure to modify all target sites. The finding that most instances of non-methylation are associated with hemi-methylation is congruent with observations that hemi-methylated palindromes are resistant to cleavage by restriction endonucleases. However, sites where hemi-methylation is conserved might represent a unique regulatory function for PBCV-1. This study serves as a baseline for future investigation into the epigenomics of chloroviruses and their giant virus relatives.
绿藻病毒(科)感染真核淡水单细胞绿藻。这些病毒的一个独特特征是大量存在DNA甲基转移酶,一些分离株将其蛋白质编码潜能的4.5%用于这些基因。这种多样性凸显了绿藻病毒模型系统的一个长期价值;在该系统中,全基因组表观基因组特征分析可能开始阐明双链DNA大病毒中DNA甲基化的功能。我们使用单分子实时(SMRT)测序(PacBio)对原型绿藻病毒PBCV-1中的DNA修饰进行了表征。将结果与仅基于DNA序列预测的总可用位点进行了比较。SMRT软件在GATC和CATG识别位点检测到N6-甲基腺嘌呤(m6A),这两个基序先前分别被证明是PBCV-1 DNA甲基转移酶M.CviAI和M.AII的作用靶点。与此同时,PacBio分析表明,PBCV-1基因组的10.9%具有较大的脉冲间隔持续时间比(ipdRatio)值,这是识别DNA修饰的主要指标。这些位点比GATC和CATG基序中所有可用腺嘌呤所能解释的位点多20.6倍,这表明可能存在除甲基化以外的碱基或骨架修饰。为了确定甲基化稳定性,我们对三个生物学重复中每个GATC和CATG序列的甲基化状态进行了交叉比较,发现约81%的位点被稳定甲基化,而约2%的位点始终缺乏甲基化。其余17%的位点是随机甲基化的。当对每个靶点的两条链的甲基化状态进行分析时,我们发现回文序列存在于完全未甲基化状态、完全甲基化状态或半甲基化状态,尽管GATC位点比CATG序列更常缺乏甲基化。鉴于这两个序列不仅是甲基转移酶的作用靶点,也是由PBCV-1作为病毒起源的限制修饰(RM)系统共同编码的限制性内切酶的作用靶点,因此存在强烈的选择压力来修饰所有靶点。大多数未甲基化情况与半甲基化相关的这一发现与半甲基化回文序列对限制性内切酶切割具有抗性的观察结果一致。然而,半甲基化保守的位点可能代表了PBCV-1的一种独特调节功能。这项研究为未来对绿藻病毒及其巨型病毒亲属的表观基因组学研究奠定了基础。