Graduate School of Advanced Science and Engineering, Waseda University, 55N-06-10, 3-4-1, Okubo Shinjuku-ku, Tokyo, 169-8555, Japan.
Computational Bio Big-Data Open Innovation Laboratory (CBBD-OIL), National Institute of Advanced Industrial Science and Technology (AIST), Tokyo, Japan.
Microbiome. 2020 Jun 23;8(1):95. doi: 10.1186/s40168-020-00864-3.
The human gut microbiome has been suggested to affect human health and thus has received considerable attention. To clarify the structure of the human gut microbiome, clustering methods are frequently applied to human gut taxonomic profiles. Enterotypes, i.e., clusters of individuals with similar microbiome composition, are well-studied and characterized. However, only a few detailed studies on assemblages, i.e., clusters of co-occurring bacterial taxa, have been conducted. Particularly, the relationship between the enterotype and assemblage is not well-understood.
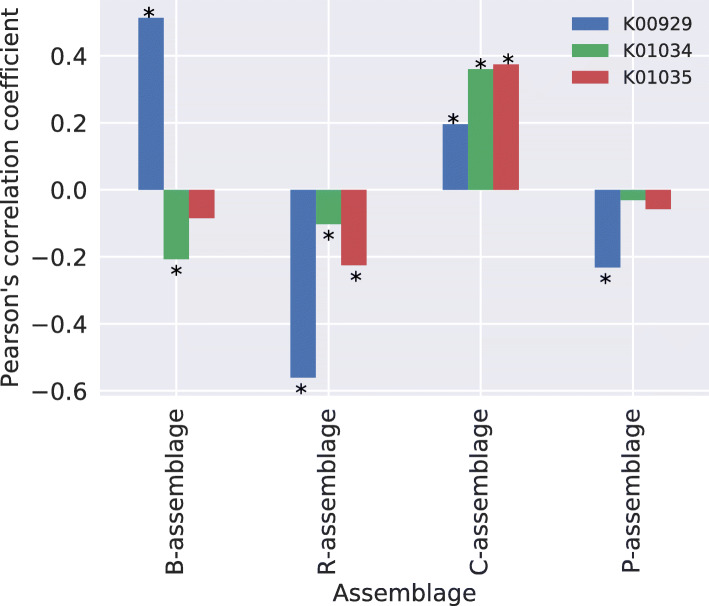
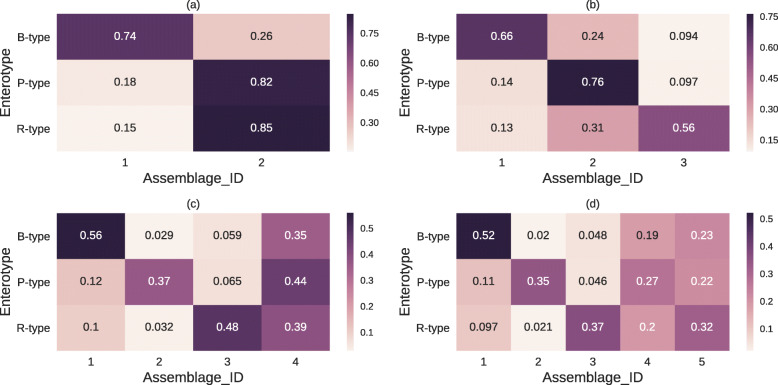
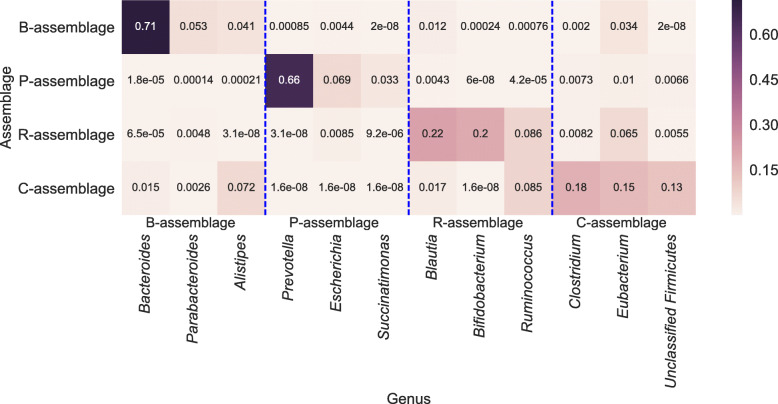
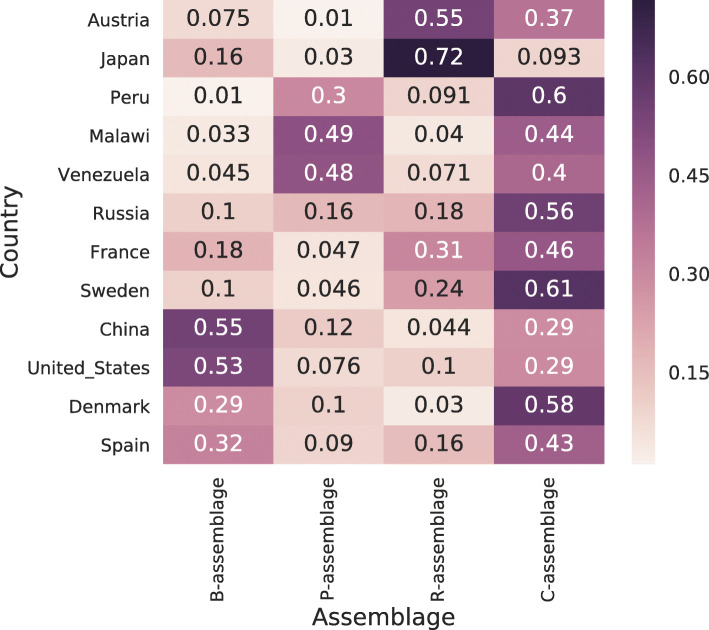
In this study, we detected gut microbiome assemblages using a latent Dirichlet allocation (LDA) method. We applied LDA to a large-scale human gut metagenome dataset and found that a 4-assemblage LDA model could represent relationships between enterotypes and assemblages with high interpretability. This model indicated that each individual tends to have several assemblages, three of which corresponded to the three classically recognized enterotypes. Conversely, the fourth assemblage corresponded to no enterotypes and emerged in all enterotypes. Interestingly, the dominant genera of this assemblage (Clostridium, Eubacterium, Faecalibacterium, Roseburia, Coprococcus, and Butyrivibrio) included butyrate-producing species such as Faecalibacterium prausnitzii. Indeed, the fourth assemblage significantly positively correlated with three butyrate-producing functions.
We conducted an assemblage analysis on a large-scale human gut metagenome dataset using LDA. The present study revealed that there is an enterotype-independent assemblage. Video Abstract.
人类肠道微生物群被认为会影响人类健康,因此受到了广泛关注。为了阐明人类肠道微生物群的结构,聚类方法经常被应用于人类肠道分类学特征。肠型,即具有相似微生物群组成的个体聚类,已经得到了很好的研究和描述。然而,只有少数关于共生体的详细研究,即共同出现的细菌分类群聚类,已经开展。特别是,肠型与共生体之间的关系还没有被很好地理解。
在本研究中,我们使用潜在狄利克雷分配(LDA)方法检测了肠道微生物群的共生体。我们将 LDA 应用于一个大规模的人类肠道宏基因组数据集,发现一个 4 共生体 LDA 模型可以用高可解释性来表示肠型和共生体之间的关系。该模型表明,每个个体通常具有几个共生体,其中三个与三个经典的肠型相对应。相反,第四个共生体与任何肠型都不对应,并且出现在所有肠型中。有趣的是,这个共生体的主要属(Clostridium、Eubacterium、Faecalibacterium、Roseburia、Coprococcus 和 Butyrivibrio)包括产丁酸的物种,如 Faecalibacterium prausnitzii。事实上,第四个共生体与三个产丁酸的功能呈显著正相关。
我们使用 LDA 对一个大规模的人类肠道宏基因组数据集进行了共生体分析。本研究揭示了存在一种与肠型无关的共生体。