Jiang Shuang, Xiao Guanghua, Koh Andrew Y, Chen Yingfei, Yao Bo, Li Qiwei, Zhan Xiaowei
Department of Statistical Science, Southern Methodist University, Dallas, TX, United States.
Quantitative Biomedical Research Center, Department of Population and Data Sciences, University of Texas Southwestern Medical Center, Dallas, TX, United States.
Front Genet. 2020 Jun 3;11:445. doi: 10.3389/fgene.2020.00445. eCollection 2020.
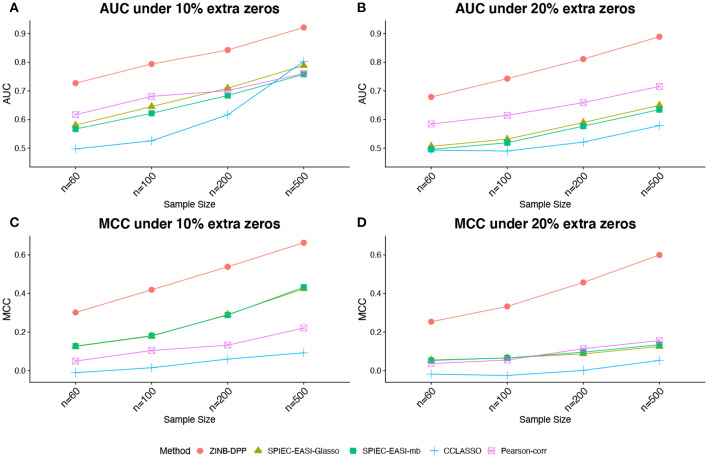
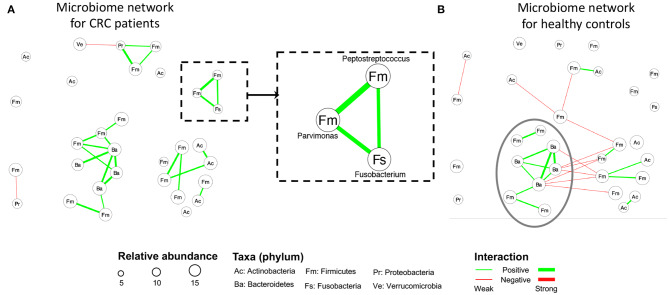
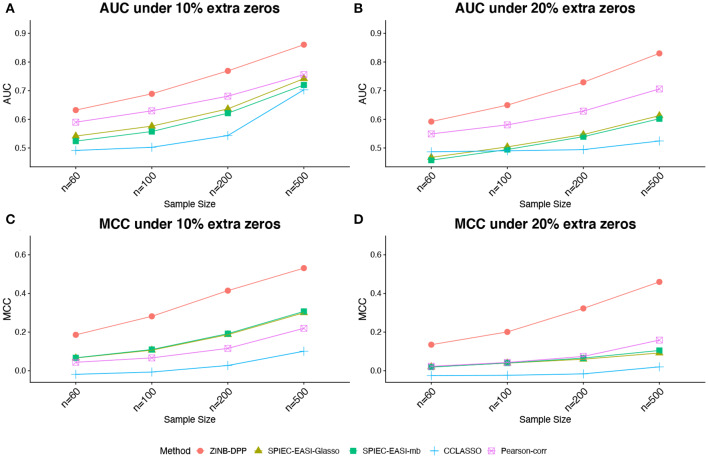
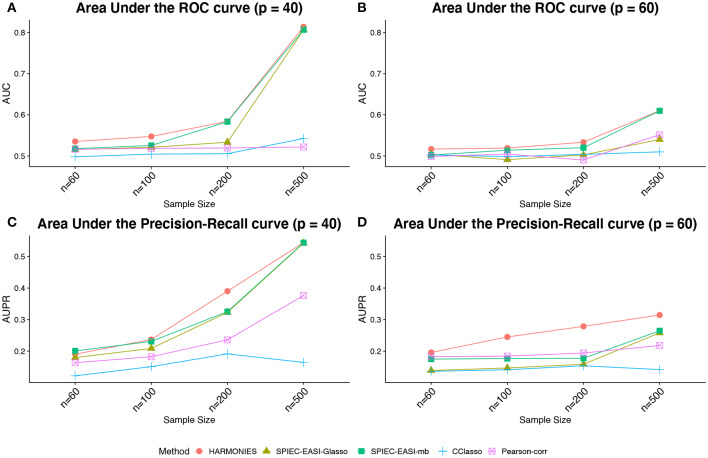
The human microbiome is a collection of microorganisms. They form complex communities and collectively affect host health. Recently, the advances in next-generation sequencing technology enable the high-throughput profiling of the human microbiome. This calls for a statistical model to construct microbial networks from the microbiome sequencing count data. As microbiome count data are high-dimensional and suffer from uneven sampling depth, over-dispersion, and zero-inflation, these characteristics can bias the network estimation and require specialized analytical tools. Here we propose a general framework, HARMONIES, Hybrid Approach foR MicrobiOme Network Inferences via Exploiting Sparsity, to infer a sparse microbiome network. HARMONIES first utilizes a zero-inflated negative binomial (ZINB) distribution to model the skewness and excess zeros in the microbiome data, as well as incorporates a stochastic process prior for sample-wise normalization. This approach infers a sparse and stable network by imposing non-trivial regularizations based on the Gaussian graphical model. In comprehensive simulation studies, HARMONIES outperformed four other commonly used methods. When using published microbiome data from a colorectal cancer study, it discovered a novel community with disease-enriched bacteria. In summary, HARMONIES is a novel and useful statistical framework for microbiome network inference, and it is available at https://github.com/shuangj00/HARMONIES.
人类微生物组是微生物的集合。它们形成复杂的群落并共同影响宿主健康。最近,下一代测序技术的进步使得对人类微生物组进行高通量分析成为可能。这就需要一个统计模型来从微生物组测序计数数据构建微生物网络。由于微生物组计数数据具有高维性,并且存在采样深度不均、过度离散和零膨胀等问题,这些特征会使网络估计产生偏差,因此需要专门的分析工具。在此,我们提出了一个通用框架HARMONIES,即通过利用稀疏性进行微生物组网络推断的混合方法,来推断稀疏的微生物组网络。HARMONIES首先利用零膨胀负二项式(ZINB)分布对微生物组数据中的偏度和过多零值进行建模,并纳入用于样本归一化的随机过程先验。该方法通过基于高斯图形模型施加非平凡正则化来推断稀疏且稳定的网络。在全面的模拟研究中,HARMONIES优于其他四种常用方法。当使用来自一项结直肠癌研究的已发表微生物组数据时,它发现了一个富含疾病相关细菌的新群落。总之,HARMONIES是一种用于微生物组网络推断的新颖且有用的统计框架,可在https://github.com/shuangj00/HARMONIES获取。