Zhao Haitao, Duan Zhong-Hui
Integrated Bioscience Program, The University of Akron, Akron, OH, USA.
Department of Computer Science, The University of Akron, Akron, OH, USA.
Bioinform Biol Insights. 2019 Apr 8;13:1177932219839402. doi: 10.1177/1177932219839402. eCollection 2019.
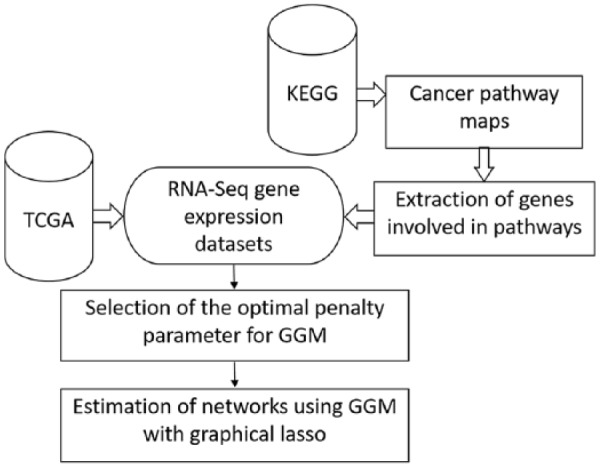
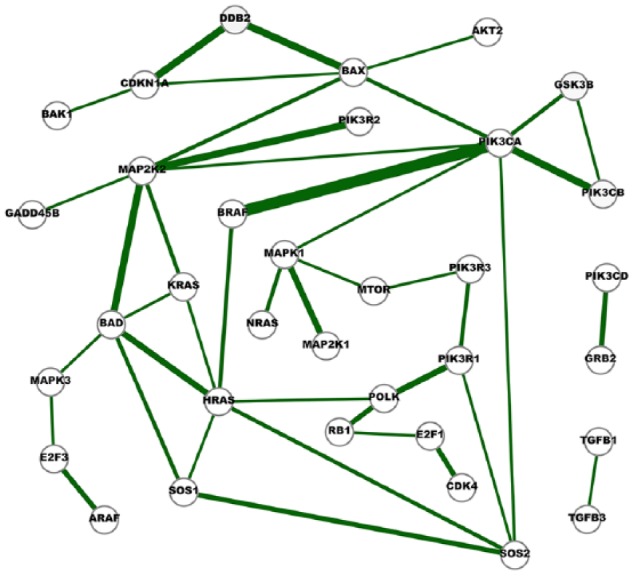
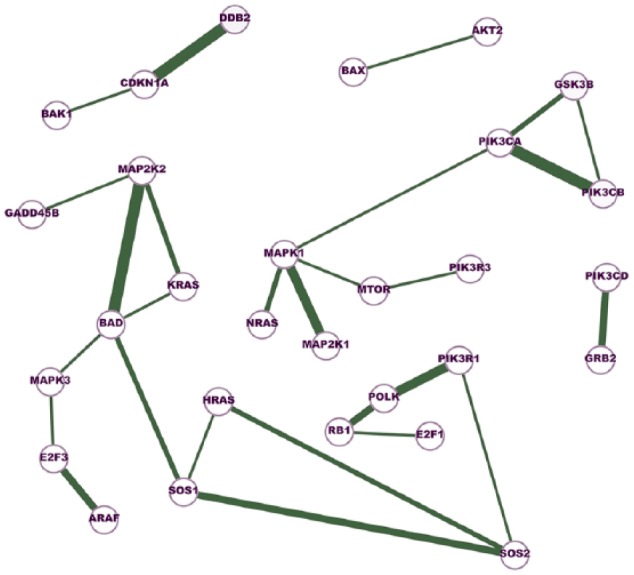
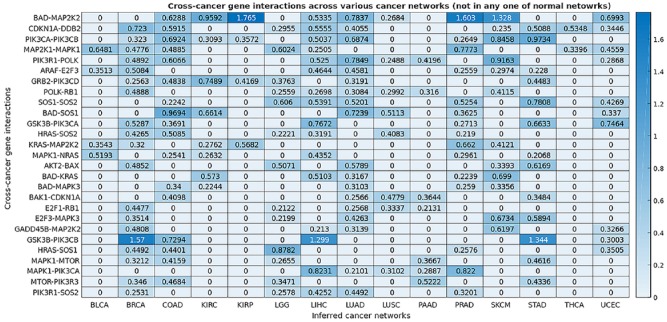
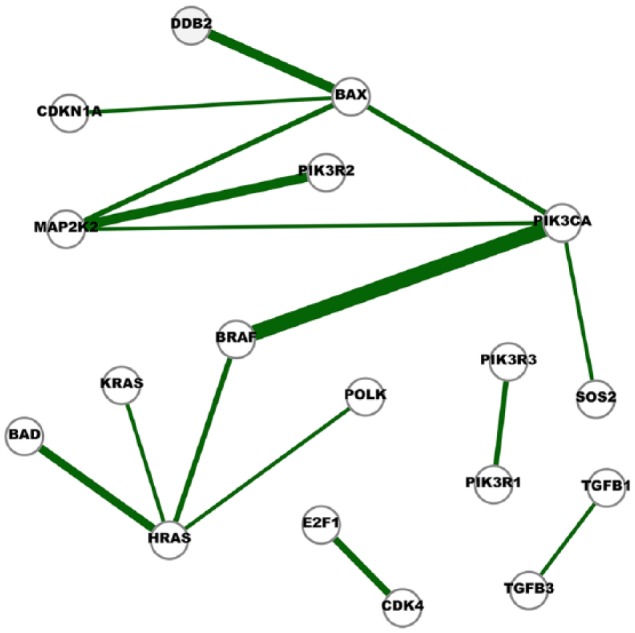
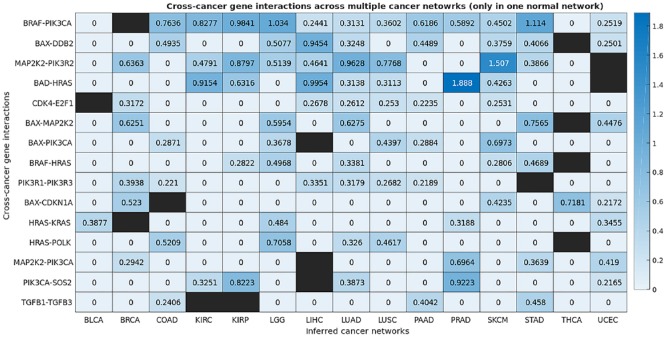
The Cancer Genome Atlas (TCGA) provides a rich resource that can be used to understand how genes interact in cancer cells and has collected RNA-Seq gene expression data for many types of human cancer. However, mining the data to uncover the hidden gene-interaction patterns remains a challenge. Gaussian graphical model (GGM) is often used to learn genetic networks because it defines an undirected graphical structure, revealing the conditional dependences of genes. In this study, we focus on inferring gene interactions in 15 specific types of human cancer using RNA-Seq expression data and GGM with graphical lasso. We take advantage of the corresponding Kyoto Encyclopedia of Genes and Genomes pathway maps to define the subsets of related genes. RNA-Seq expression levels of the subsets of genes in solid cancerous tumor and normal tissues were extracted from TCGA. The gene expression data sets were cleaned and formatted, and the genetic network corresponding to each cancer type was then inferred using GGM with graphical lasso. The inferred networks reveal stable conditional dependences among the genes at the expression level and confirm the essential roles played by the genes that encode proteins involved in the two key signaling pathway phosphoinositide 3-kinase (PI3K)/AKT/mTOR and Ras/Raf/MEK/ERK in human carcinogenesis. These stable dependences elucidate the expression level interactions among the genes that are implicated in many different human cancers. The inferred genetic networks were examined to further identify and characterize a collection of gene interactions that are unique to cancer. The cross-cancer genetic interactions revealed from our study provide another set of knowledge for cancer biologists to propose strong hypotheses, so further biological investigations can be conducted effectively.
癌症基因组图谱(TCGA)提供了丰富的资源,可用于了解基因在癌细胞中的相互作用方式,并且已经收集了多种人类癌症的RNA测序基因表达数据。然而,挖掘这些数据以发现隐藏的基因相互作用模式仍然是一项挑战。高斯图形模型(GGM)常用于学习遗传网络,因为它定义了一种无向图形结构,揭示了基因的条件依赖性。在本研究中,我们专注于使用RNA测序表达数据和带图形套索的GGM来推断15种特定类型人类癌症中的基因相互作用。我们利用相应的京都基因与基因组百科全书通路图来定义相关基因的子集。从TCGA中提取实体癌肿瘤和正常组织中基因子集的RNA测序表达水平。对基因表达数据集进行清理和格式化,然后使用带图形套索的GGM推断每种癌症类型对应的遗传网络。推断出的网络揭示了基因在表达水平上稳定的条件依赖性,并证实了编码参与两个关键信号通路磷酸肌醇3激酶(PI3K)/AKT/mTOR和Ras/Raf/MEK/ERK的蛋白质的基因在人类致癌过程中所起的重要作用。这些稳定的依赖性阐明了涉及多种不同人类癌症的基因之间的表达水平相互作用。对推断出的遗传网络进行检查,以进一步识别和表征一组癌症特有的基因相互作用。我们的研究揭示的跨癌症遗传相互作用为癌症生物学家提出有力假设提供了另一套知识,以便能够有效地进行进一步的生物学研究。