GLAZgo Discovery Centre, Institute of Infection, Immunity & Inflammation, College of Medicine, Veterinary and Life Sciences, University of Glasgow, Glasgow, United Kingdom.
Centre for Immunobiology, Institute of Infection, Immunity & Inflammation, College of Medicine, Veterinary and Life Sciences, University of Glasgow, Glasgow, United Kingdom.
J Biol Chem. 2020 Aug 14;295(33):11754-11763. doi: 10.1074/jbc.RA120.014113. Epub 2020 Jun 25.
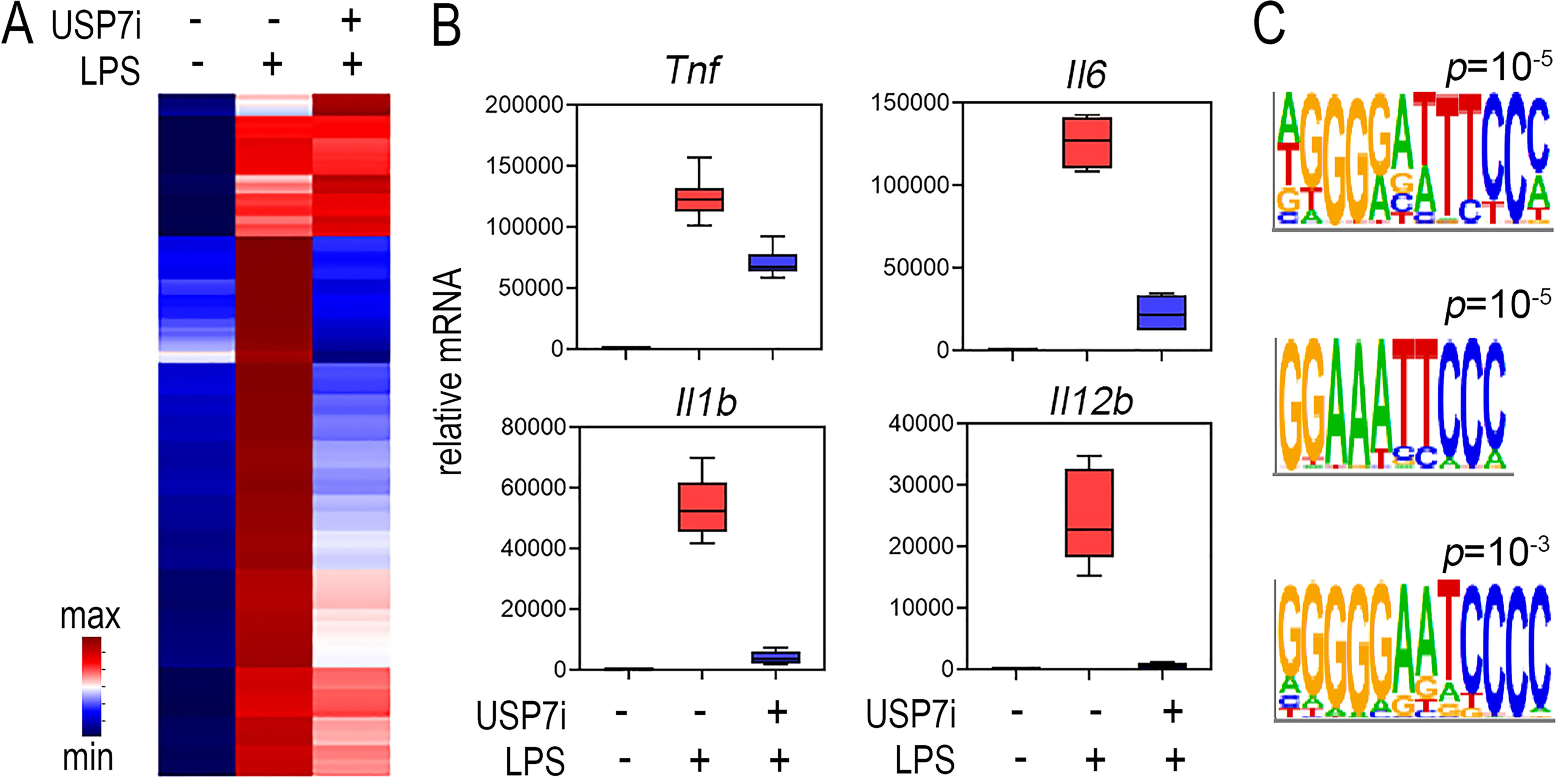
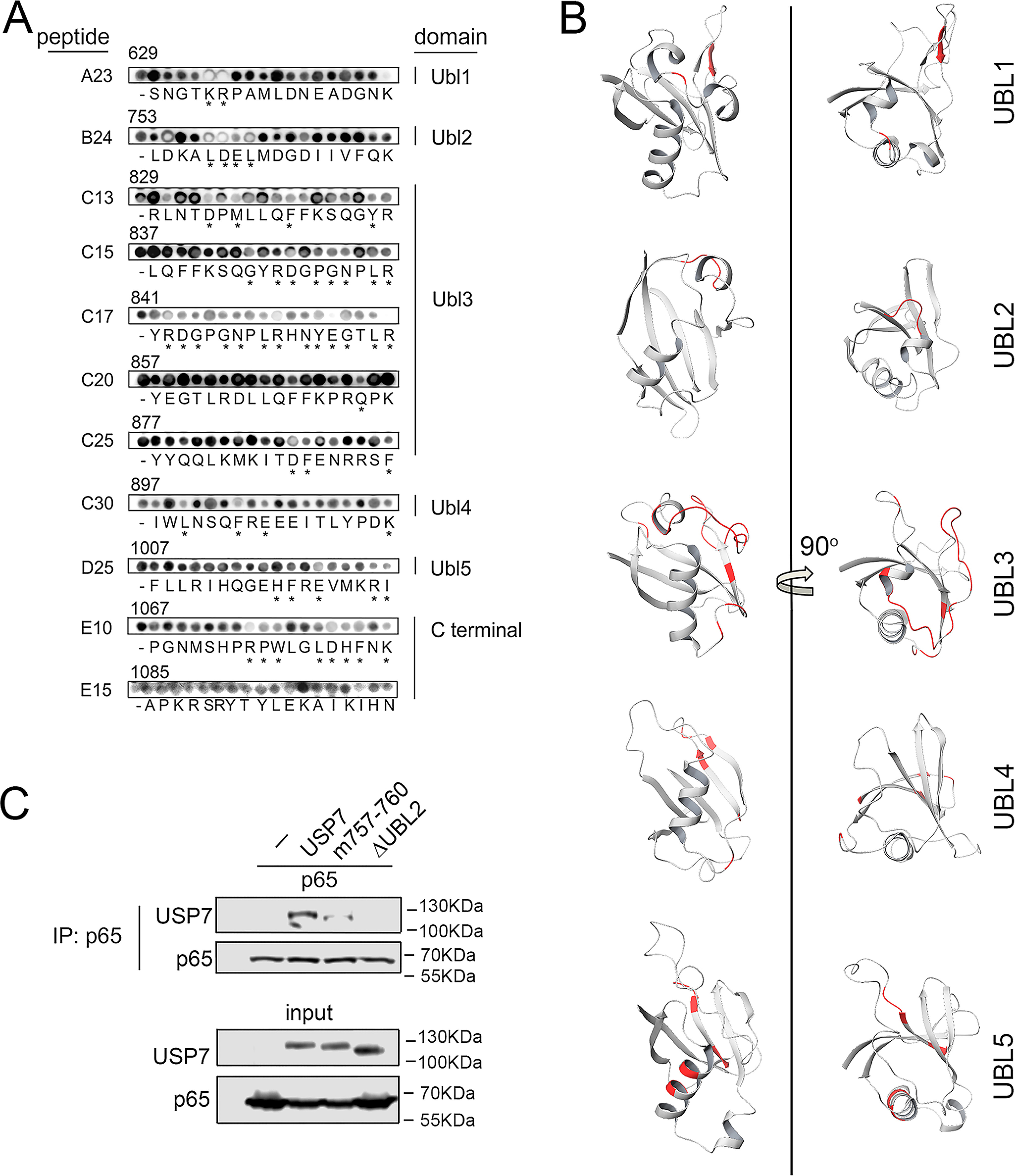
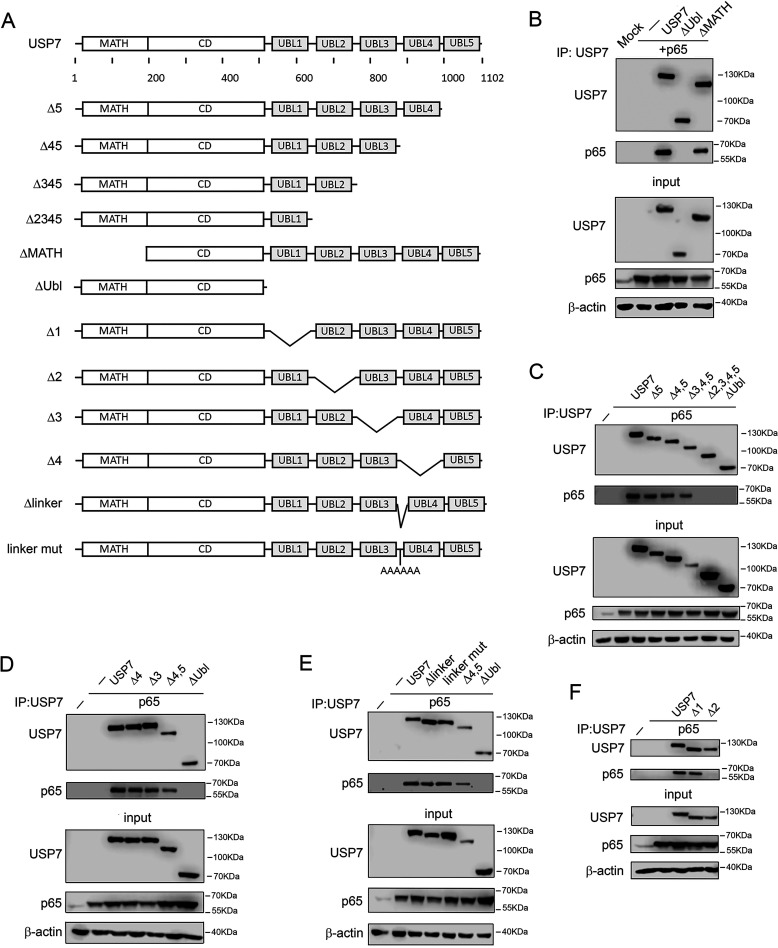
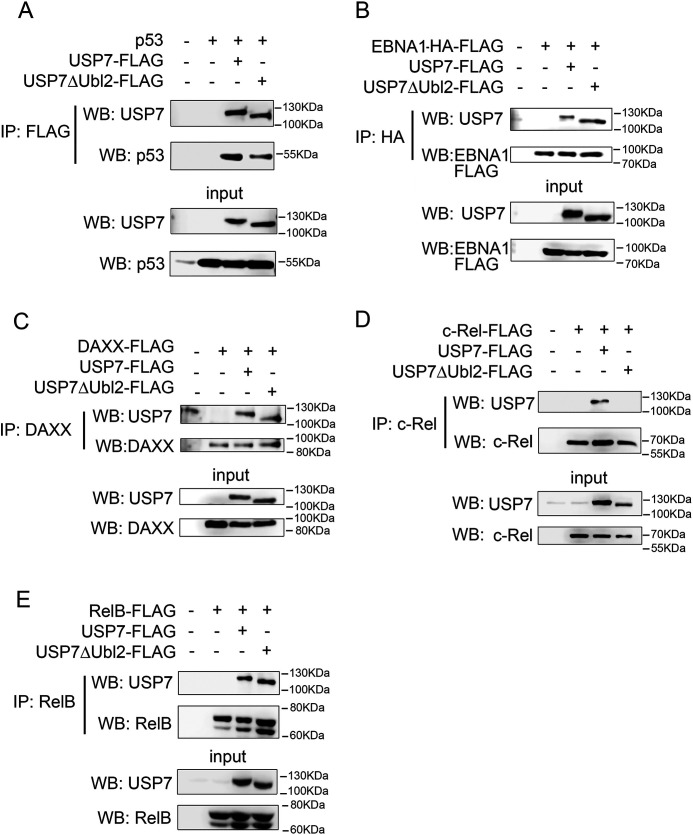
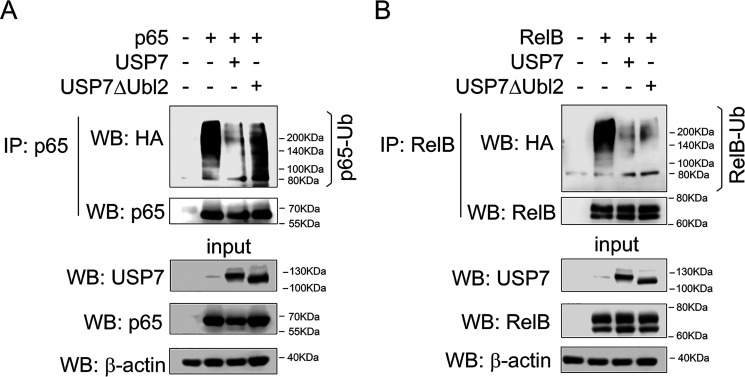
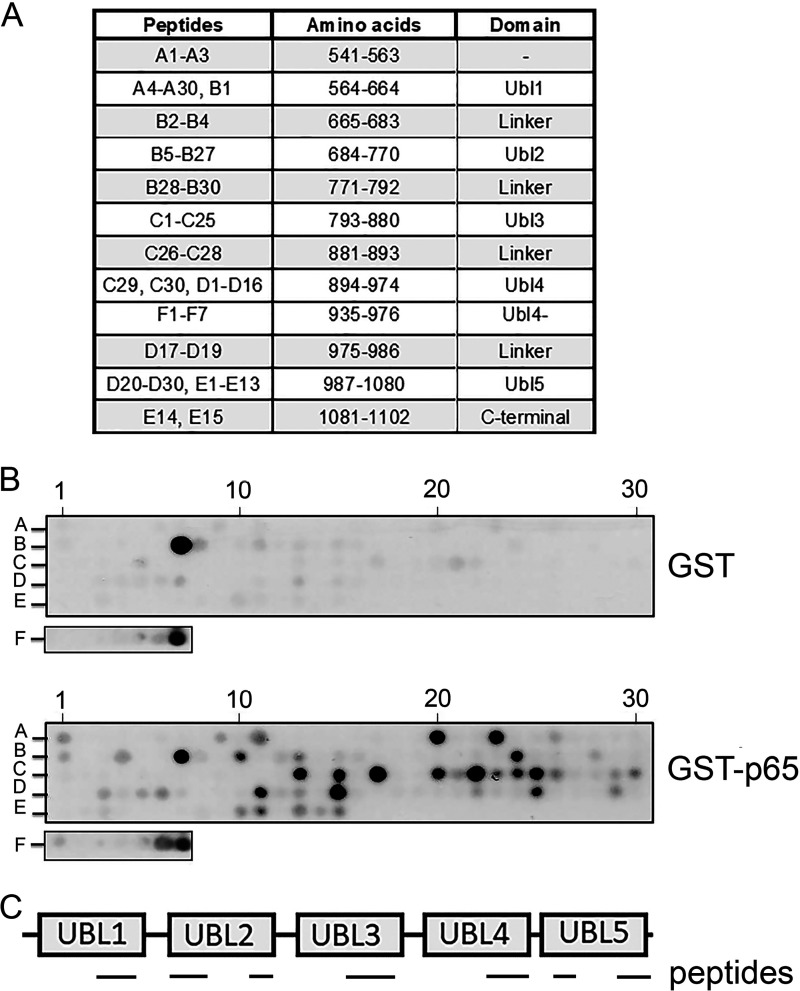
The transcription factor NF-ĸB is a master regulator of the innate immune response and plays a central role in inflammatory diseases by mediating the expression of pro-inflammatory cytokines. Ubiquitination-triggered proteasomal degradation of DNA-bound NF-ĸB strongly limits the expression of its target genes. Conversely, USP7 (deubiquitinase ubiquitin-specific peptidase 7) opposes the activities of E3 ligases, stabilizes DNA-bound NF-ĸB, and thereby promotes NF-ĸB-mediated transcription. Using gene expression and synthetic peptide arrays on membrane support and overlay analyses, we found here that inhibiting USP7 increases NF-ĸB ubiquitination and degradation, prevents Toll-like receptor-induced pro-inflammatory cytokine expression, and represents an effective strategy for controlling inflammation. However, the broad regulatory roles of USP7 in cell death pathways, chromatin, and DNA damage responses limit the use of catalytic inhibitors of USP7 as anti-inflammatory agents. To this end, we identified an NF-ĸB-binding site in USP7, ubiquitin-like domain 2, that selectively mediates interactions of USP7 with NF-ĸB subunits but is dispensable for interactions with other proteins. Moreover, we found that the amino acids LDEL in USP7 critically contribute to the interaction with the p65 subunit of NF-ĸB. Our findings support the notion that USP7 activity could be potentially targeted in a substrate-selective manner through the development of noncatalytic inhibitors of this deubiquitinase to abrogate NF-ĸB activity.
转录因子 NF-κB 是先天免疫反应的主要调节因子,通过介导促炎细胞因子的表达,在炎症性疾病中发挥核心作用。泛素化触发的 NF-κB 蛋白水解体降解强烈限制了其靶基因的表达。相反,USP7(去泛素化酶泛素特异性肽酶 7)拮抗 E3 连接酶的活性,稳定与 DNA 结合的 NF-κB,并促进 NF-κB 介导的转录。我们在此使用膜支持和覆盖分析的基因表达和合成肽阵列,发现抑制 USP7 可增加 NF-κB 的泛素化和降解,防止 Toll 样受体诱导的促炎细胞因子表达,是控制炎症的有效策略。然而,USP7 在细胞死亡途径、染色质和 DNA 损伤反应中的广泛调节作用限制了 USP7 的催化抑制剂作为抗炎剂的使用。为此,我们在 USP7 的泛素样结构域 2 中鉴定出一个 NF-κB 结合位点,该位点选择性介导 USP7 与 NF-κB 亚基的相互作用,但对于与其他蛋白质的相互作用是可有可无的。此外,我们发现 USP7 中的氨基酸 LDEL 对于与 NF-κB 的 p65 亚基的相互作用至关重要。我们的研究结果支持了这样一种观点,即 USP7 的活性可以通过开发该去泛素酶的非催化抑制剂,以特定于底物的方式进行靶向,从而阻断 NF-κB 的活性。