Department Sanitat i Anatomia Animals, Facultat de Veterinària, Universitat Autònoma de Barcelona, Travessera dels Turons s/n, campus UAB, Cerdanyola del Vallès, Spain.
Ceva Salud Animal, S.A. Avenida Diagonal, Barcelona, Spain.
Transbound Emerg Dis. 2021 Mar;68(2):519-530. doi: 10.1111/tbed.13709. Epub 2020 Jul 14.
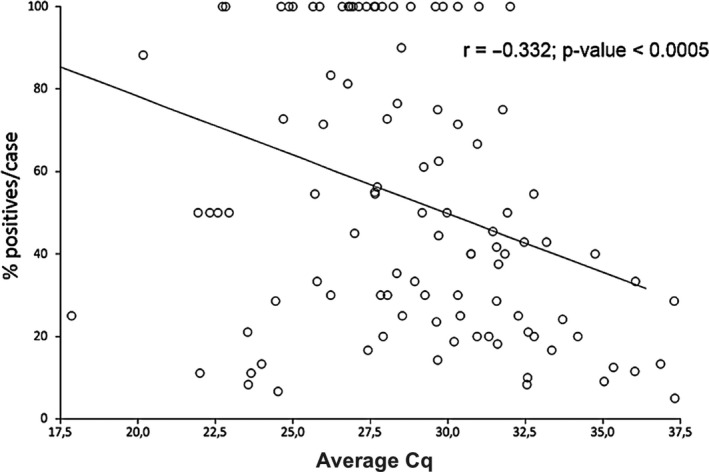
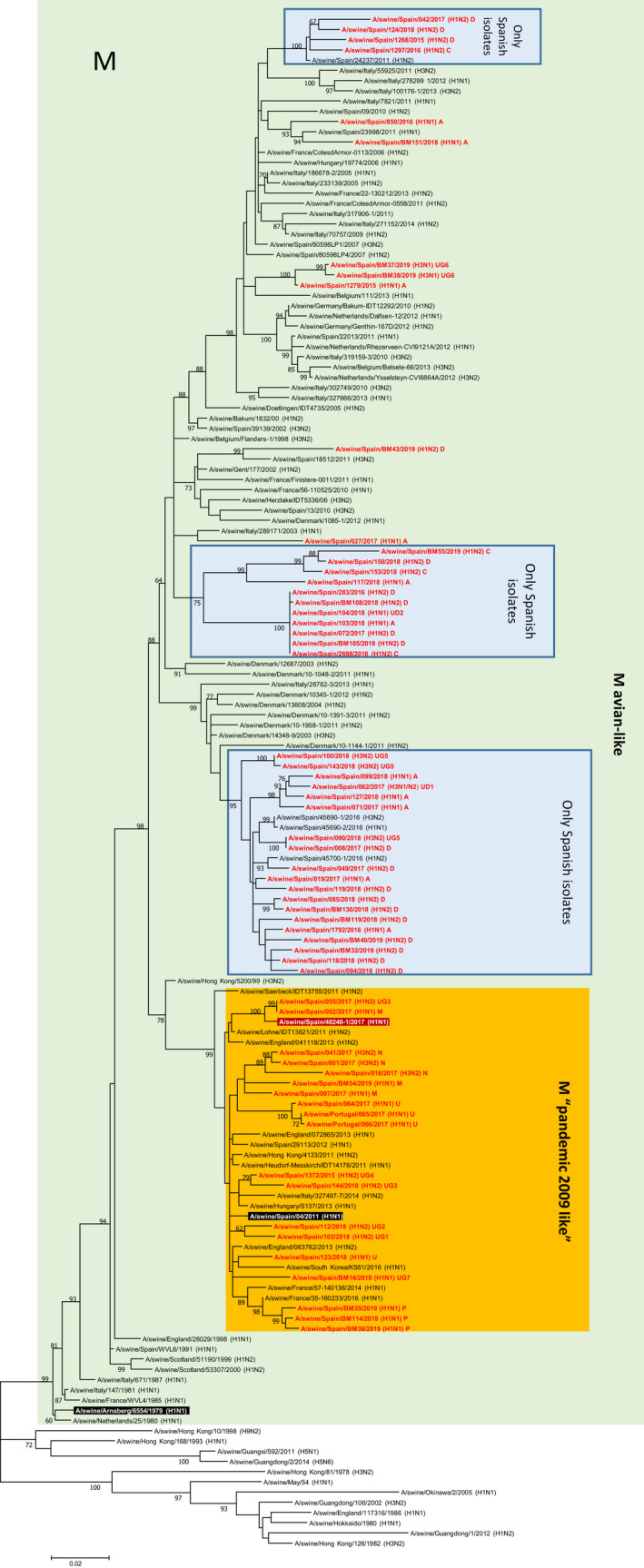
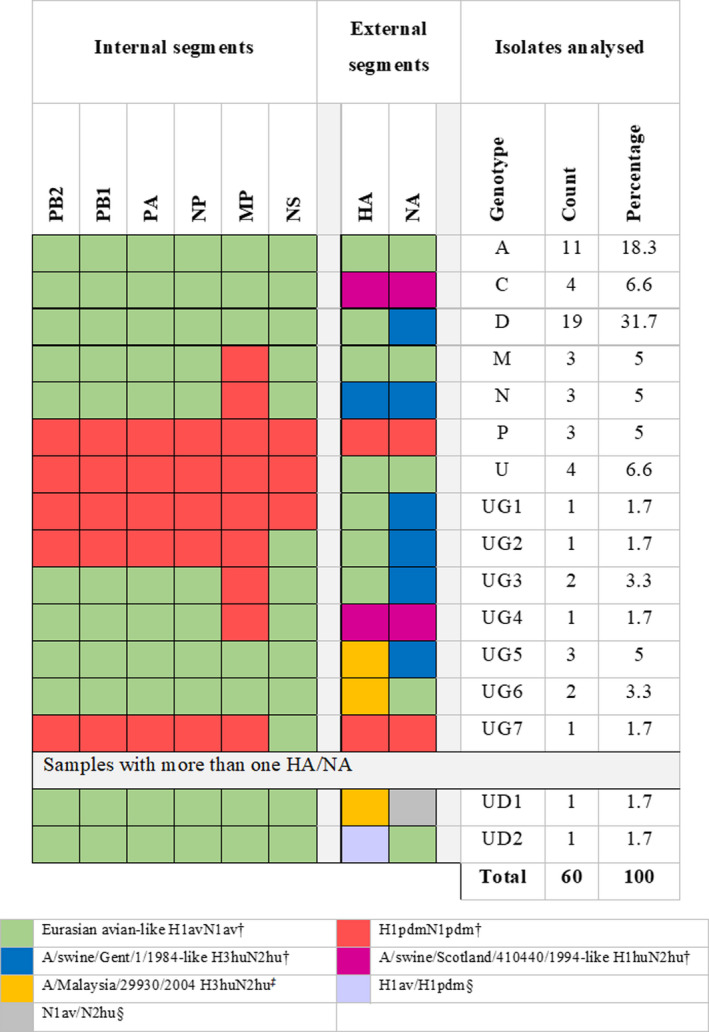
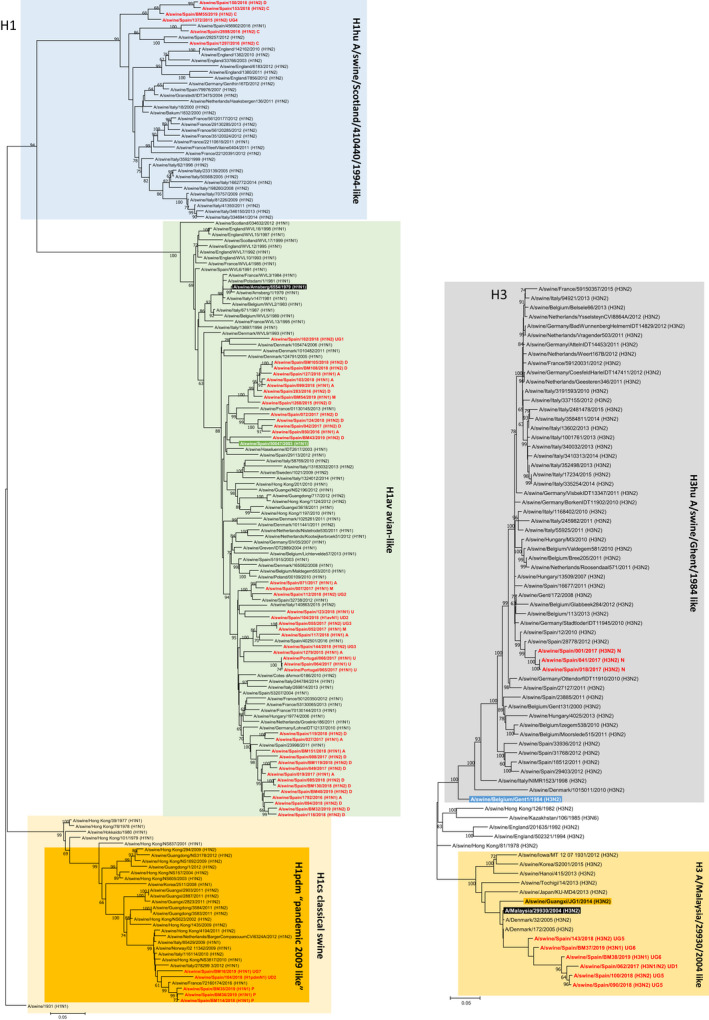
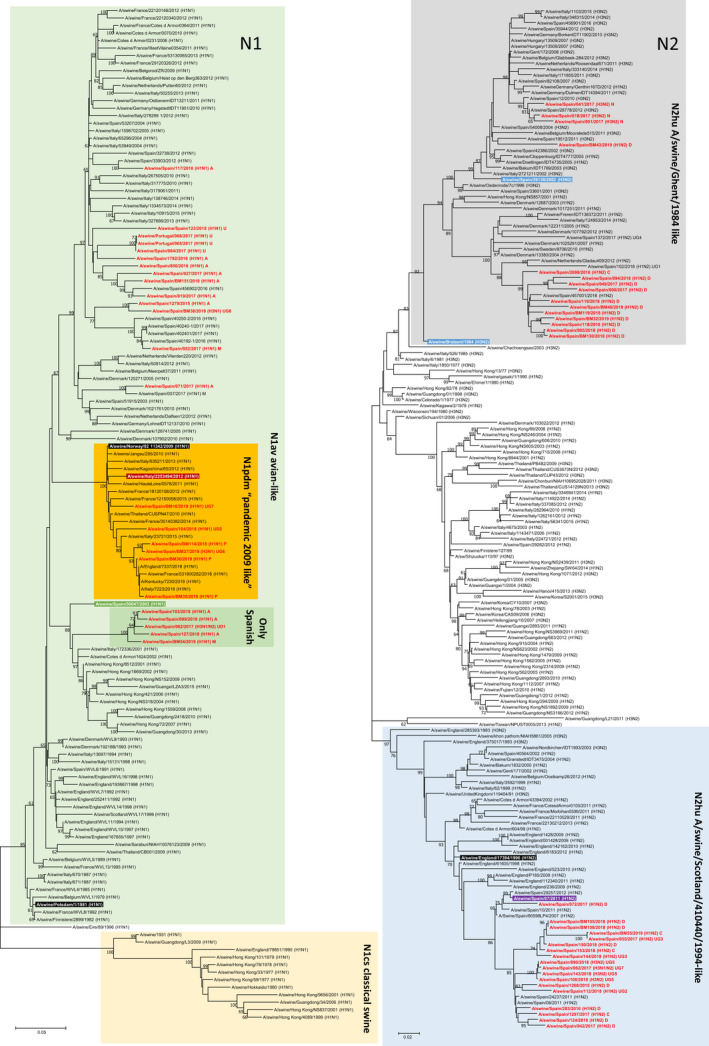
The present study was aimed to assess the diversity of influenza A viruses (IAV) circulating in pig farms in the Iberian Peninsula. The study included two different situations: farms suffering respiratory disease outbreaks compatible with IAV (n = 211) and randomly selected farms without overt respiratory disease (n = 19). Initially, the presence of IAV and lineage determination was assessed by qRT-PCR using nasal swabs. IAV was confirmed in 145 outbreaks (68.7%), mostly in nurseries (53/145; 36.5%). Subtyping by RT-qPCR was possible in 94 of those cases being H1avN2hu (33.6%), H1avN1av (24.3%) and H1huN2hu (18.7%), the most common lineages. H3huN2hu and H1pdmN1pdm represented 7.5% and 6.5% of the cases, respectively. As for the randomly selected farms, 15/19 (78.9%) were positive for IAV. Again, the virus was mostly found in nurseries and H1avN2hu was the predominant lineage. Virus isolation in MDCK cells was attempted from positive cases. Sixty of the isolates were fully sequenced with Illumina MiSeq®. Within those 60 isolates, the most frequent genotypes had internal genes of avian origin, and these were D (19/60; 31.7%) and A (11/60; 18.3%), H1avN2hu and H1avN1av, respectively. In addition, seven previously unreported genotypes were identified. In two samples, more than one H or N were found and it was not possible to precisely establish their genotypes. A great diversity was observed in the phylogenetic analysis. Notably, four H3 sequences clustered with human isolates from 2004-05 (Malaysia and Denmark) that were considered uncommon in pigs. Overall, this study indicates that IAV is a very common agent in respiratory disease outbreaks in Spanish pig farms. The genetic diversity of this virus is continuously expanding with clear changes in the predominant subtypes and lineages in relatively short periods of time. The current genotyping scheme has to be enlarged to include the new genotypes that could be found in the future.
本研究旨在评估伊比利亚半岛养猪场中流行的甲型流感病毒 (IAV) 的多样性。该研究包括两种不同情况:出现与 IAV 相符的呼吸道疾病暴发的农场(n=211)和随机选择的无明显呼吸道疾病的农场(n=19)。最初,通过使用鼻拭子进行 qRT-PCR 评估了 IAV 的存在和谱系确定。在 145 次暴发(68.7%)中确认了 IAV,主要发生在育肥舍(53/145;36.5%)。在这些情况下,通过 RT-qPCR 进行的亚型分析显示,H1avN2hu(33.6%)、H1avN1av(24.3%)和 H1huN2hu(18.7%)是最常见的谱系。H3huN2hu 和 H1pdmN1pdm 分别占病例的 7.5%和 6.5%。至于随机选择的农场,19/19(78.9%)的 IAV 呈阳性。同样,病毒主要存在于育肥舍中,H1avN2hu 是主要的谱系。尝试从阳性病例的 MDCK 细胞中分离病毒。用 Illumina MiSeq®对 60 株分离株进行了全序列测序。在这 60 个分离株中,最常见的基因型具有禽类来源的内部基因,分别为 D(19/60;31.7%)和 A(11/60;18.3%)、H1avN2hu 和 H1avN1av。此外,还鉴定了七种以前未报道的基因型。在两个样本中发现了一个以上的 H 或 N,无法准确确定其基因型。在系统发育分析中观察到了很大的多样性。值得注意的是,有四个 H3 序列与 2004-05 年(马来西亚和丹麦)的人类分离株聚类,这些分离株在猪中被认为不常见。总的来说,这项研究表明,IAV 是西班牙养猪场呼吸道疾病暴发的一种非常常见的病原体。该病毒的遗传多样性不断扩大,主要亚型和谱系在相对较短的时间内发生明显变化。目前的基因分型方案必须扩大,以包括未来可能发现的新基因型。