Shanghai Clinical Center for Endocrine and Metabolic Diseases, Shanghai Institute of Endocrine and Metabolic Diseases, Department of Endocrine and Metabolic Diseases, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, 200025, China.
School of Medicine, Cheeloo College of Medicine, Shandong University; Center for Reproductive Medicine, Cheeloo College of Medicine, Shandong University, Jinan, Shandong, 250012, China.
Theranostics. 2020 Jun 5;10(16):7351-7368. doi: 10.7150/thno.44459. eCollection 2020.
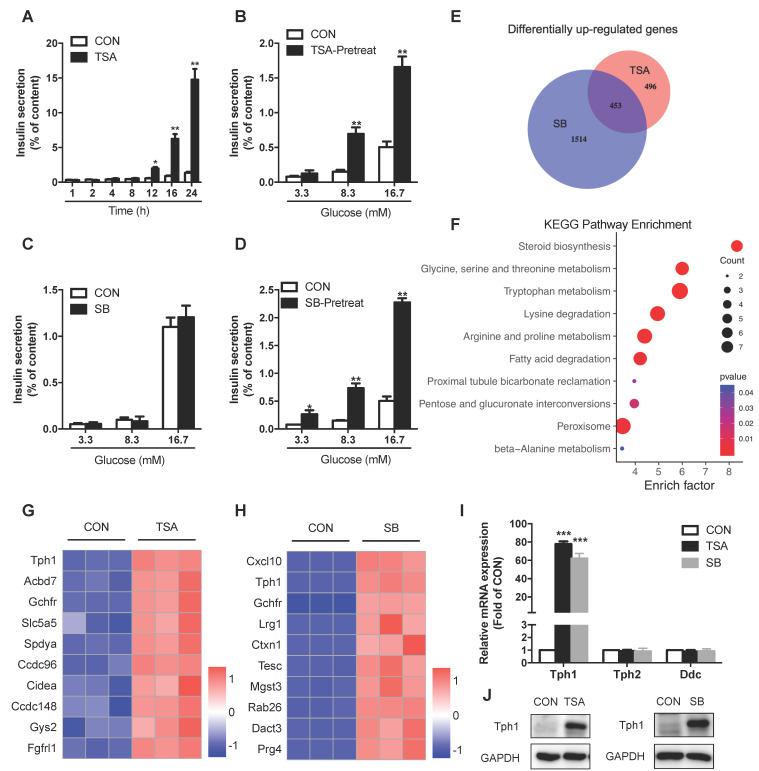
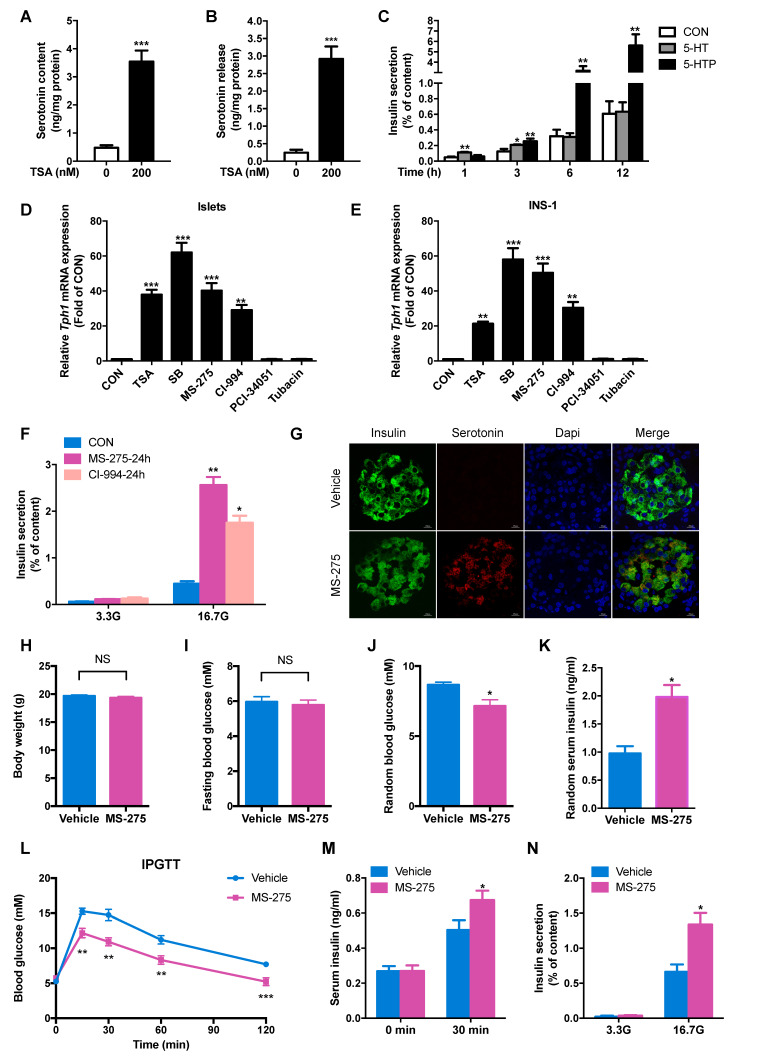
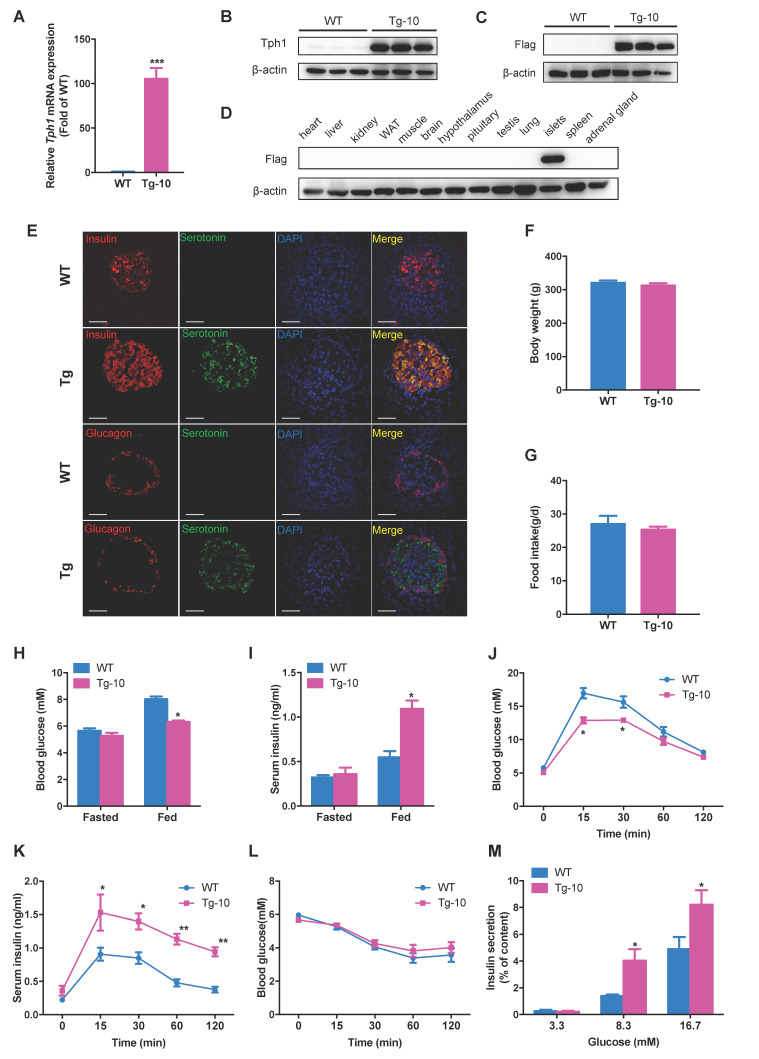
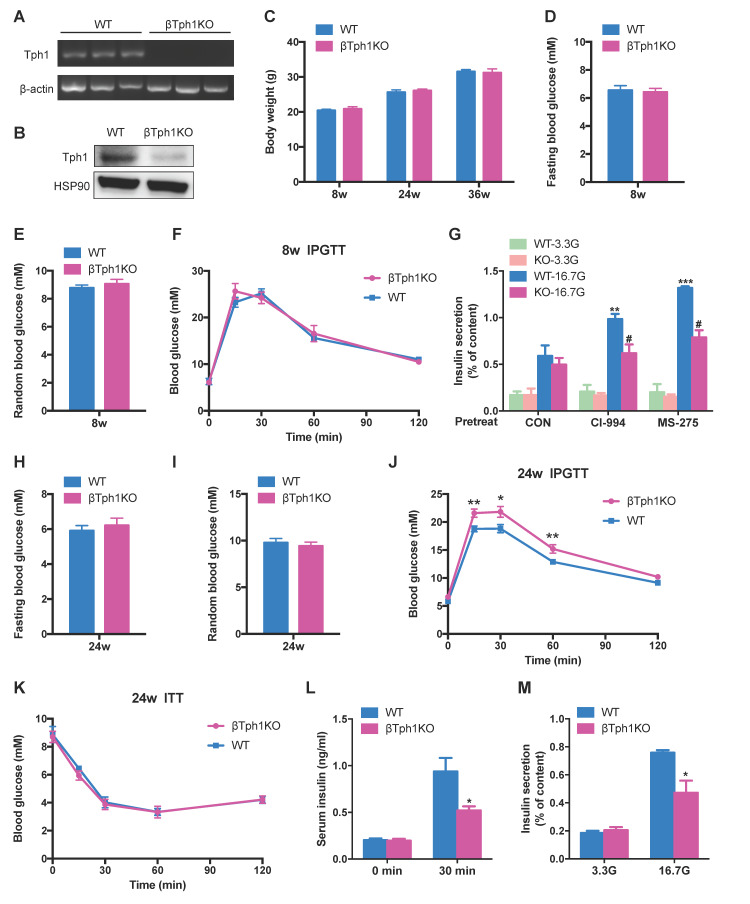
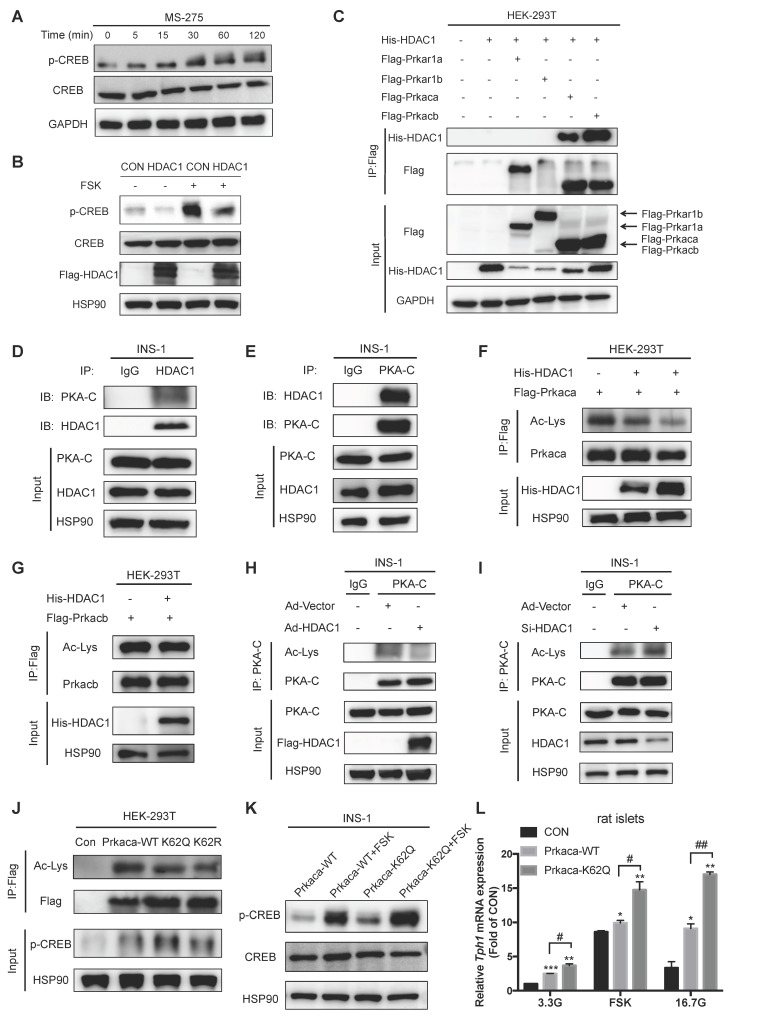
Protein acetylation is tightly linked to transcriptional control and energy metabolism. However, the role of protein acetylation in islet function remains enigmatic. This study aims to determine how protein acetylation controls β-cell function and explore the underlying mechanism. The gene-expression profiles were analyzed for rat islets in response to two histone deacetylase (HDAC) inhibitors. Insulin secretion, tryptophan hydroxylase 1 (Tph1) expression, and serotonin synthesis of rat islets were detected after HDAC inhibitor treatment both and β-cell-specific Tph1-overexpressing transgenic rats and β-cell-specific Tph1 knockout mice were constructed to evaluate the role of Tph1 in β-cell function. The deacetylation of PKA in β-cells by HDAC1 was investigated by adenoviral infection, immunoprecipitation, and western blot. Inhibition of HDACs greatly potentiated pancreatic β-cell function and reprogrammed transcriptional landscape of islets. Among the commonly up-regulated genes by two pan-HDAC inhibitors, displayed the most prominent change. Specifically, inhibition of HDAC1 and HDAC3 by MS-275 strongly promoted Tph1 expression and endogenous serotonin synthesis in rat islets, concomitantly with enhanced insulin secretory capacity and β-cell-specific Tph1-overexpressing transgenic rats exhibited improved glucose tolerance and amplified glucose-stimulated insulin secretion. On the contrary, β-cell-specific Tph1 knockout mice displayed glucose intolerance and impaired insulin secretion with aging. Moreover, depletion of Tph1 in β-cells abrogated MS-275-induced insulin hypersecretion. Overexpression of HDAC1, not HDAC3, inhibited Tph1 transcriptional activity and decreased MS-275-stimulated Tph1 expression. Mechanistically, HDAC1 deacetylated PKA catalytic subunit and decreased its activity, resulting in Tph1 transcriptional repression. The acetylation mimetic K62Q mutant of PKA increased its catalytic activity. HDAC1 inhibition exerted a synergistic effect with cAMP/PKA signal on Tph1 expression. The present findings highlight a novel role of HDAC1-PKA-Tph1 signaling in governing β-cell functional compensation by derepressing serotonin synthesis.
蛋白质乙酰化与转录控制和能量代谢密切相关。然而,蛋白质乙酰化在胰岛功能中的作用仍然是个谜。本研究旨在确定蛋白质乙酰化如何控制β细胞功能,并探讨其潜在机制。
分析了两种组蛋白去乙酰化酶(HDAC)抑制剂作用下大鼠胰岛的基因表达谱。用 HDAC 抑制剂处理大鼠胰岛后,检测胰岛素分泌、色氨酸羟化酶 1(Tph1)表达和 5-羟色胺合成。构建了胰岛β细胞特异性 Tph1 过表达转基因大鼠和β细胞特异性 Tph1 敲除小鼠,以评估 Tph1 在β细胞功能中的作用。通过腺病毒感染、免疫沉淀和 Western blot 研究了 HDAC1 对β细胞中 PKA 的去乙酰化作用。
HDACs 的抑制极大地增强了胰腺β细胞的功能,并重塑了胰岛的转录谱。在两种泛 HDAC 抑制剂共同上调的基因中,显示出最显著的变化。具体来说,MS-275 强烈抑制 HDAC1 和 HDAC3,可促进大鼠胰岛 Tph1 表达和内源性 5-羟色胺合成,同时增强胰岛素分泌能力。胰岛β细胞特异性 Tph1 过表达转基因大鼠表现出改善的葡萄糖耐量和增强的葡萄糖刺激胰岛素分泌。相反,随着年龄的增长,β细胞特异性 Tph1 敲除小鼠表现出葡萄糖不耐受和胰岛素分泌受损。此外,β细胞中 Tph1 的耗竭消除了 MS-275 诱导的胰岛素分泌过度。HDAC1 的过表达而非 HDAC3 的过表达抑制了 Tph1 的转录活性并降低了 MS-275 刺激的 Tph1 表达。机制上,HDAC1 使 PKA 催化亚基去乙酰化并降低其活性,导致 Tph1 转录抑制。PKA 的乙酰化模拟物 K62Q 突变体增加其催化活性。HDAC1 抑制与 cAMP/PKA 信号对 Tph1 表达具有协同作用。
本研究结果强调了 HDAC1-PKA-Tph1 信号在通过去抑制 5-羟色胺合成来调节β细胞功能补偿方面的新作用。