Chen Jian-Min, Lin Jin-Huan, Masson Emmanuelle, Liao Zhuan, Férec Claude, Cooper David N, Hayden Matthew
1EFS, Univ Brest, Inserm, UMR 1078, GGB, F-29200Brest, France; 2Department of Gastroenterology, Changhai Hospital, Second Military Medical University, Shanghai, China; 3Shanghai Institute of Pancreatic Diseases, Shanghai, China; 4CHRU Brest, Service de Génétique Médicale et de Biologie de la Reproduction, Brest, France; 5Institute of Medical Genetics, School of Medicine, Cardiff University, Cardiff, UK.
Curr Genomics. 2020 Jan;21(1):56-66. doi: 10.2174/1389202921666200210141701.
5' splice site GT>GC or +2T>C variants have been frequently reported to cause human genetic disease and are routinely scored as pathogenic splicing mutations. However, we have recently demonstrated that such variants in human disease genes may not invariably be pathogenic. Moreover, we found that no splicing prediction tools appear to be capable of reliably distinguishing those +2T>C variants that generate wild-type transcripts from those that do not.
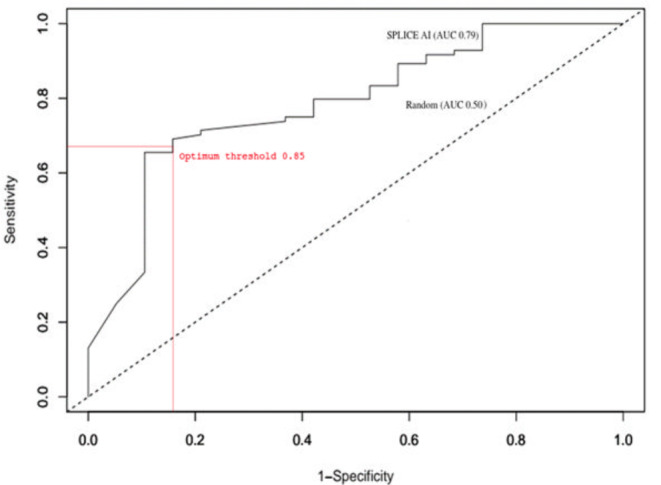
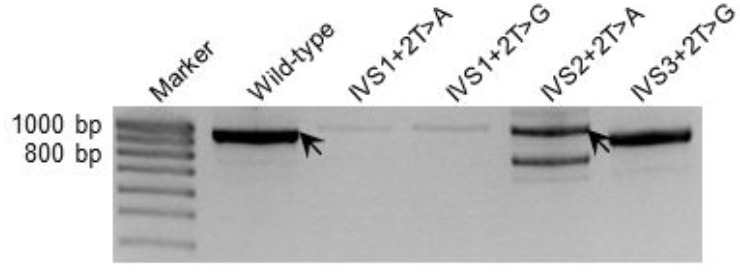
Herein, we evaluated the performance of a novel deep learning-based tool, SpliceAI, in the context of three datasets of +2T>C variants, all of which had been characterized functionally in terms of their impact on pre-mRNA splicing. The first two datasets refer to our recently described "" dataset of 45 known disease-causing +2T>C variants and the "" dataset of 103 +2T>C substitutions subjected to full-length gene splicing assay. The third dataset comprised 12 +2T>C variants that were recently analyzed by saturation genome editing.
Comparison of the SpliceAI-predicted and experimentally obtained functional impact assessments of these variants (and smaller datasets of +2T>A and +2T>G variants) revealed that although SpliceAI performed rather better than other prediction tools, it was still far from perfect. A key issue was that the impact of those +2T>C (and +2T>A) variants that generated wild-type transcripts represents a quantitative change that can vary from barely detectable to an almost full expression of wild-type transcripts, with wild-type transcripts often co-existing with aberrantly spliced transcripts.
Our findings highlight the challenges that we still face in attempting to accurately identify splice-altering variants.
5'剪接位点GT>GC或+2T>C变异体经常被报道会导致人类遗传疾病,并且通常被判定为致病性剪接突变。然而,我们最近证明,人类疾病基因中的此类变异体不一定总是致病性的。此外,我们发现没有一种剪接预测工具似乎能够可靠地区分那些产生野生型转录本的+2T>C变异体和不产生野生型转录本的变异体。
在此,我们在三个+2T>C变异体数据集的背景下评估了一种基于深度学习的新型工具SpliceAI的性能,所有这些数据集在对前体mRNA剪接的影响方面都已进行了功能表征。前两个数据集指的是我们最近描述的45个已知致病+2T>C变异体的“”数据集和103个经过全长基因剪接分析的+2T>C替换的“”数据集。第三个数据集包含最近通过饱和基因组编辑分析的12个+2T>C变异体。
对这些变异体(以及+2T>A和+2T>G变异体的较小数据集)的SpliceAI预测和实验获得的功能影响评估进行比较后发现,尽管SpliceAI的表现比其他预测工具要好,但仍远非完美。一个关键问题是,那些产生野生型转录本的+2T>C(和+2T>A)变异体的影响代表了一种定量变化,其范围可以从几乎检测不到到野生型转录本几乎完全表达,野生型转录本通常与异常剪接的转录本共存。
我们的研究结果突出了我们在试图准确识别剪接改变变异体时仍然面临的挑战。