Department of intelligent science and technology, School of Automation, Northwestern Polytechnical University, Xi'an 710072, China.
Department of Biostatistics, Epidemiology and Informatics, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA 19104, USA.
Bioinformatics. 2020 Jul 1;36(Suppl_1):i371-i379. doi: 10.1093/bioinformatics/btaa434.
Brain imaging genetics studies the complex associations between genotypic data such as single nucleotide polymorphisms (SNPs) and imaging quantitative traits (QTs). The neurodegenerative disorders usually exhibit the diversity and heterogeneity, originating from which different diagnostic groups might carry distinct imaging QTs, SNPs and their interactions. Sparse canonical correlation analysis (SCCA) is widely used to identify bi-multivariate genotype-phenotype associations. However, most existing SCCA methods are unsupervised, leading to an inability to identify diagnosis-specific genotype-phenotype associations.
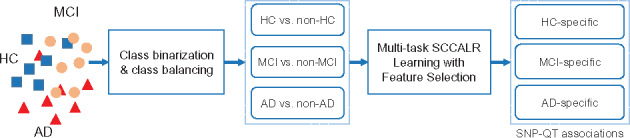
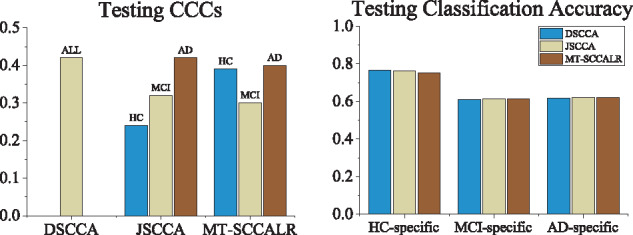
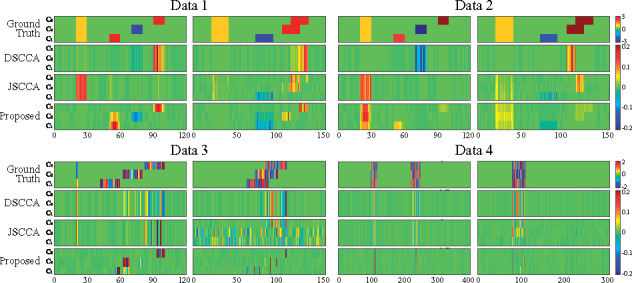
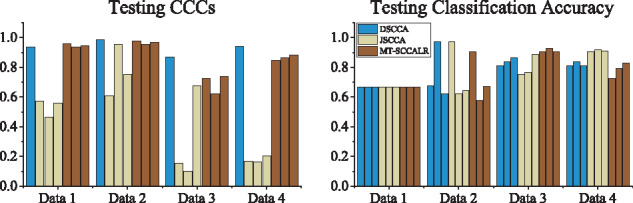
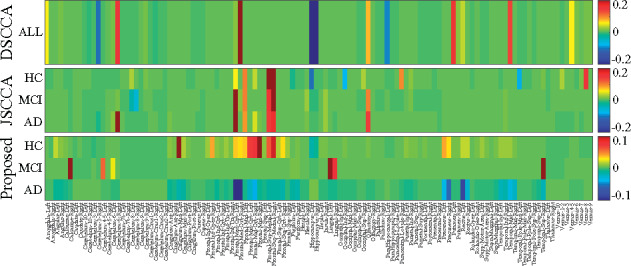
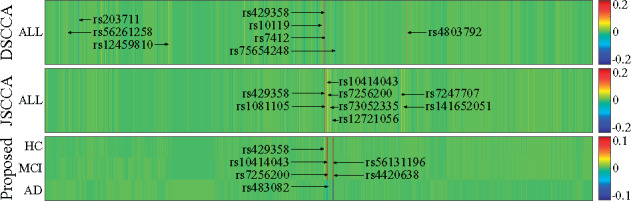
In this article, we propose a new joint multitask learning method, named MT-SCCALR, which absorbs the merits of both SCCA and logistic regression. MT-SCCALR learns genotype-phenotype associations of multiple tasks jointly, with each task focusing on identifying one diagnosis-specific genotype-phenotype pattern. Meanwhile, MT-SCCALR cannot only select relevant SNPs and imaging QTs for each diagnostic group alone, but also allows the selection of those shared by multiple diagnostic groups. We derive an efficient optimization algorithm whose convergence to a local optimum is guaranteed. Compared with two state-of-the-art methods, MT-SCCALR yields better or similar canonical correlation coefficients and classification performances. In addition, it owns much better discriminative canonical weight patterns of great interest than competitors. This demonstrates the power and capability of MTSCCAR in identifying diagnostically heterogeneous genotype-phenotype patterns, which would be helpful to understand the pathophysiology of brain disorders.
The software is publicly available at https://github.com/dulei323/MTSCCALR.
Supplementary data are available at Bioinformatics online.
脑影像遗传学研究基因型数据(如单核苷酸多态性 [SNP])与影像定量性状(QT)之间的复杂关联。神经退行性疾病通常表现出多样性和异质性,不同的诊断组可能具有不同的影像 QT、SNP 及其相互作用。稀疏典型相关分析(SCCA)广泛用于识别双多变量基因型-表型关联。然而,大多数现有的 SCCA 方法都是无监督的,导致无法识别特定诊断的基因型-表型关联。
在本文中,我们提出了一种新的联合多任务学习方法,名为 MT-SCCALR,它吸收了 SCCA 和逻辑回归的优点。MT-SCCALR 联合学习多个任务的基因型-表型关联,每个任务都专注于识别一个特定诊断的基因型-表型模式。同时,MT-SCCALR 不仅可以单独为每个诊断组选择相关的 SNP 和影像 QT,还可以允许选择多个诊断组共享的 SNP 和影像 QT。我们推导了一个有效的优化算法,保证了其收敛到局部最优解。与两种最先进的方法相比,MT-SCCALR 产生了更好或相似的典型相关系数和分类性能。此外,它还拥有比竞争对手更好的、具有重要区分力的典型权重模式。这证明了 MT-SCCALR 识别诊断异质基因型-表型模式的能力和能力,这有助于理解大脑疾病的病理生理学。
该软件可在 https://github.com/dulei323/MTSCCALR 上公开获得。
补充数据可在生物信息学在线获得。