Department of Computer Science, Princeton University, Princeton, NJ 08544, USA.
Bioinformatics. 2020 Jul 1;36(Suppl_1):i186-i193. doi: 10.1093/bioinformatics/btaa449.
Recent single-cell DNA sequencing technologies enable whole-genome sequencing of hundreds to thousands of individual cells. However, these technologies have ultra-low sequencing coverage (<0.5× per cell) which has limited their use to the analysis of large copy-number aberrations (CNAs) in individual cells. While CNAs are useful markers in cancer studies, single-nucleotide mutations are equally important, both in cancer studies and in other applications. However, ultra-low coverage sequencing yields single-nucleotide mutation data that are too sparse for current single-cell analysis methods.
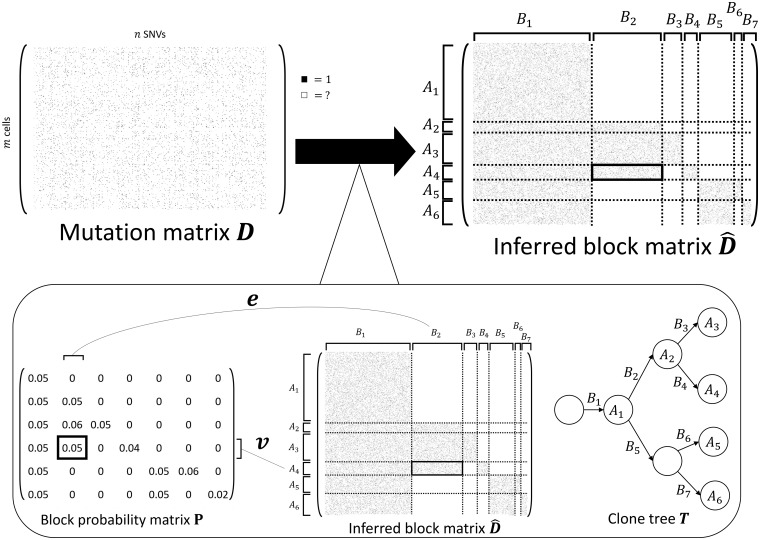
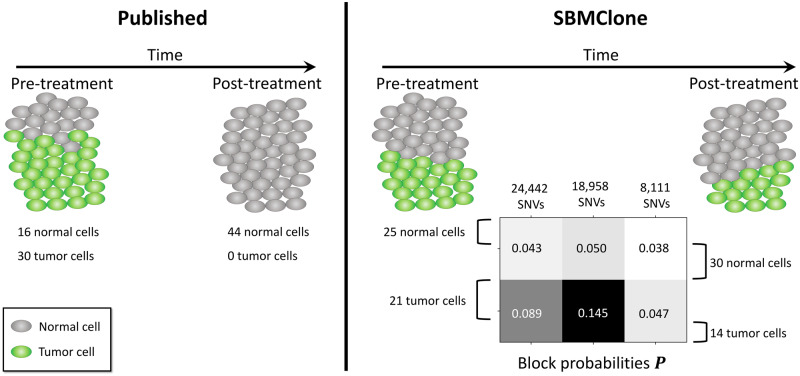
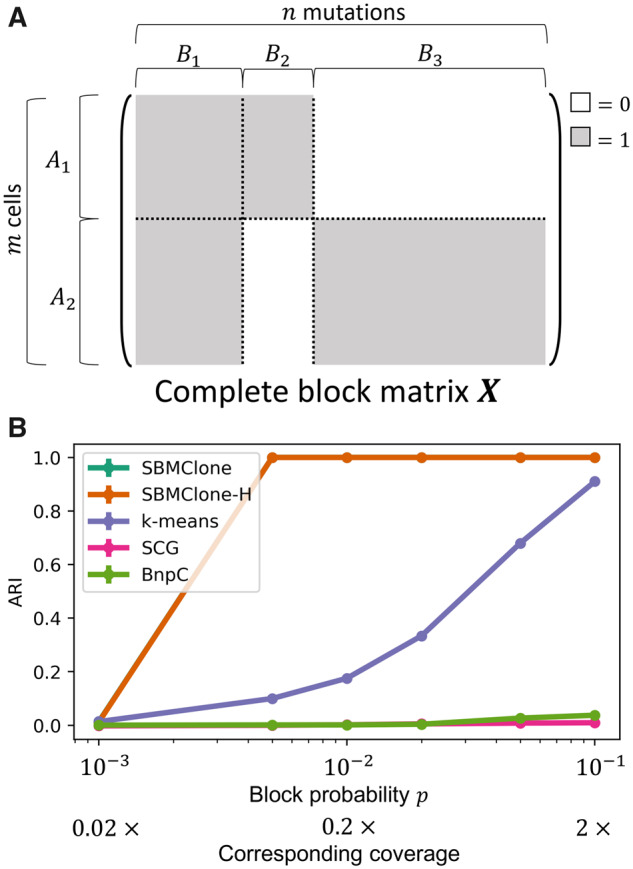
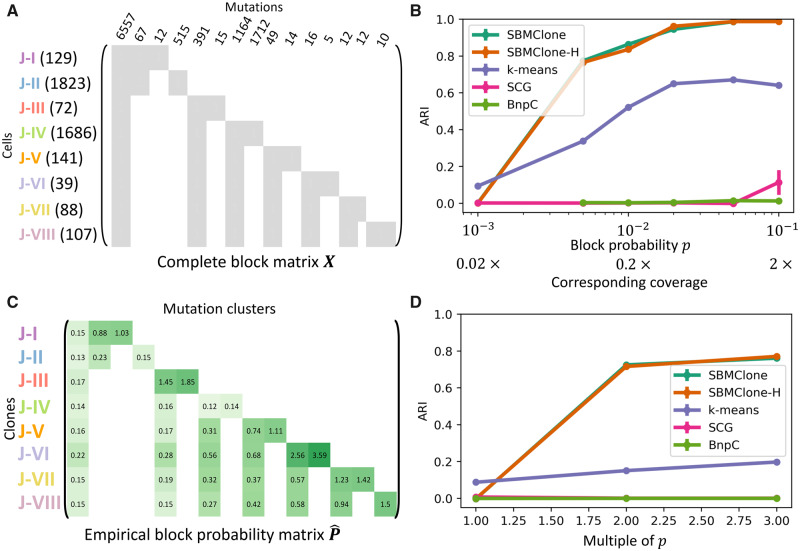
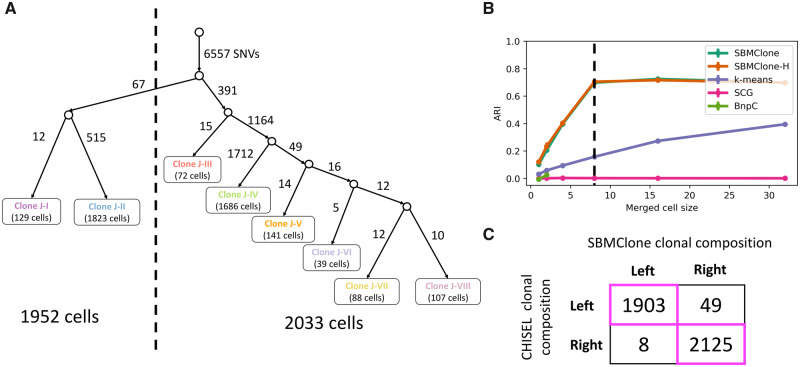
We introduce SBMClone, a method to infer clusters of cells, or clones, that share groups of somatic single-nucleotide mutations. SBMClone uses a stochastic block model to overcome sparsity in ultra-low coverage single-cell sequencing data, and we show that SBMClone accurately infers the true clonal composition on simulated datasets with coverage at low as 0.2×. We applied SBMClone to single-cell whole-genome sequencing data from two breast cancer patients obtained using two different sequencing technologies. On the first patient, sequenced using the 10X Genomics CNV solution with sequencing coverage ≈0.03×, SBMClone recovers the major clonal composition when incorporating a small amount of additional information. On the second patient, where pre- and post-treatment tumor samples were sequenced using DOP-PCR with sequencing coverage ≈0.5×, SBMClone shows that tumor cells are present in the post-treatment sample, contrary to published analysis of this dataset.
SBMClone is available on the GitHub repository https://github.com/raphael-group/SBMClone.
Supplementary data are available at Bioinformatics online.
最近的单细胞 DNA 测序技术能够对数百到数千个单个细胞进行全基因组测序。然而,这些技术的测序覆盖度超低(每个细胞<0.5×),这限制了它们在单个细胞中大拷贝数异常(CNA)分析中的应用。虽然 CNA 是癌症研究中的有用标记物,但单核苷酸突变在癌症研究和其他应用中同样重要。然而,超低覆盖度测序产生的单核苷酸突变数据对于当前的单细胞分析方法来说过于稀疏。
我们引入了 SBMClone 方法,用于推断共享体细胞单核苷酸突变群的细胞簇或克隆。SBMClone 使用随机块模型来克服超低覆盖度单细胞测序数据的稀疏性,我们表明 SBMClone 可以在覆盖度低至 0.2×的模拟数据集上准确推断真实的克隆组成。我们将 SBMClone 应用于从两名乳腺癌患者获得的两种不同测序技术的单细胞全基因组测序数据。在第一个患者中,使用 10X Genomics CNV 解决方案进行测序,测序覆盖度约为 0.03×,当纳入少量额外信息时,SBMClone 恢复了主要的克隆组成。在第二个患者中,使用 DOP-PCR 对治疗前后的肿瘤样本进行测序,测序覆盖度约为 0.5×,SBMClone 表明肿瘤细胞存在于治疗后的样本中,与该数据集的已发表分析结果相反。
SBMClone 可在 GitHub 存储库 https://github.com/raphael-group/SBMClone 上获得。
补充数据可在生物信息学在线获得。