Department of Human Genetics, University of Utah, 15 S 2030 E, Salt Lake City, UT, 84112, USA.
Base2 Genomics, LLC, Salt Lake City, UT, 84105, USA.
Genome Med. 2020 Jul 14;12(1):62. doi: 10.1186/s13073-020-00761-2.
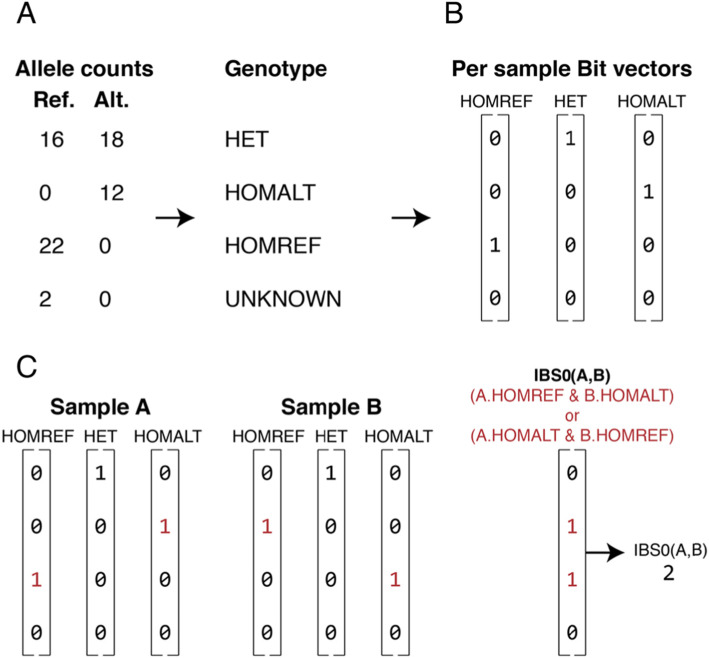
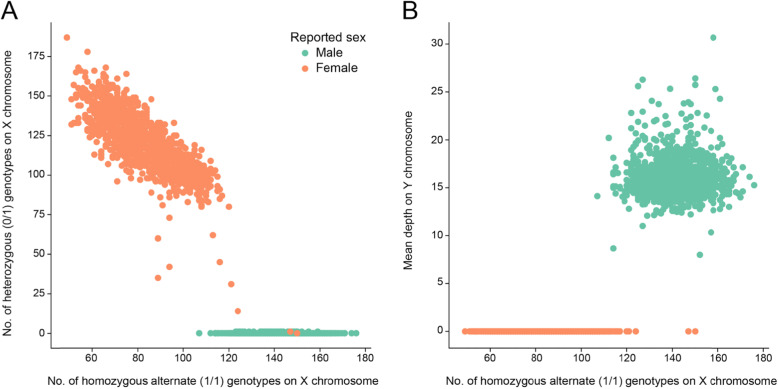
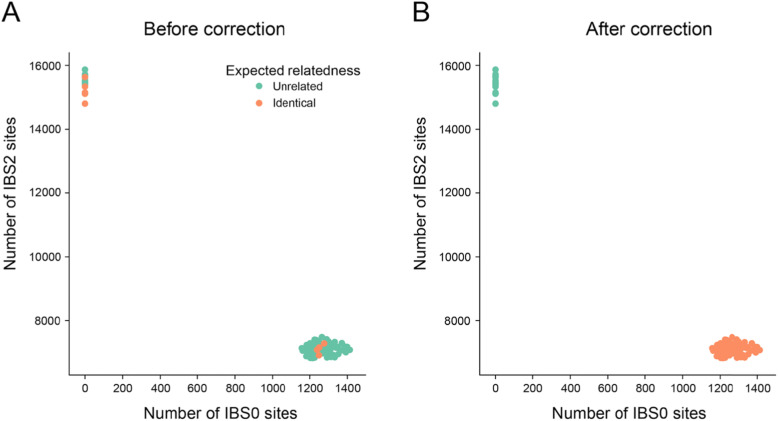
When interpreting sequencing data from multiple spatial or longitudinal biopsies, detecting sample mix-ups is essential, yet more difficult than in studies of germline variation. In most genomic studies of tumors, genetic variation is detected through pairwise comparisons of the tumor and a matched normal tissue from the sample donor. In many cases, only somatic variants are reported, which hinders the use of existing tools that detect sample swaps solely based on genotypes of inherited variants. To address this problem, we have developed Somalier, a tool that operates directly on alignments and does not require jointly called germline variants. Instead, Somalier extracts a small sketch of informative genetic variation for each sample. Sketches from hundreds of germline or somatic samples can then be compared in under a second, making Somalier a useful tool for measuring relatedness in large cohorts. Somalier produces both text output and an interactive visual report that facilitates the detection and correction of sample swaps using multiple relatedness metrics.
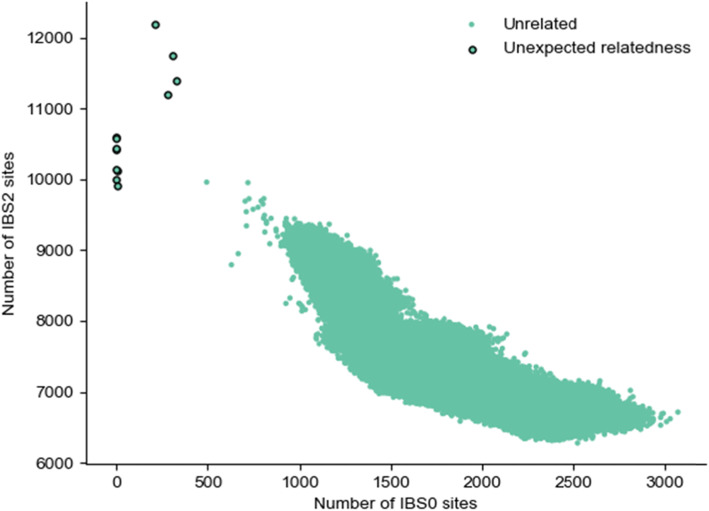
We introduce the tool and demonstrate its utility on a cohort of five glioma samples each with a normal, tumor, and cell-free DNA sample. Applying Somalier to high-coverage sequence data from the 1000 Genomes Project also identifies several related samples. We also demonstrate that it can distinguish pairs of whole-genome and RNA-seq samples from the same individuals in the Genotype-Tissue Expression (GTEx) project.
Somalier is a tool that can rapidly evaluate relatedness from sequencing data. It can be applied to diverse sequencing data types and genome builds and is available under an MIT license at github.com/brentp/somalier .
在解释来自多个空间或纵向活检的测序数据时,检测样本混淆至关重要,但比检测种系变异更困难。在大多数肿瘤的基因组研究中,通过比较肿瘤和样本供体的匹配正常组织来检测遗传变异。在许多情况下,仅报告体细胞变异,这阻碍了使用仅基于遗传变异基因型检测样本交换的现有工具。为了解决这个问题,我们开发了 Somalier,这是一种直接在比对上运行的工具,不需要共同调用种系变体。相反,Somalier 从每个样本中提取一小部分有信息的遗传变异。然后可以在不到一秒的时间内比较数百个种系或体细胞样本的草图,这使得 Somalier 成为在大队列中测量相关性的有用工具。Somalier 生成文本输出和交互式可视化报告,这有助于使用多种相关性指标检测和纠正样本交换。
我们介绍了该工具,并在一个包含五个胶质瘤样本的队列中展示了其实用性,每个样本都有一个正常、肿瘤和无细胞 DNA 样本。在 1000 基因组计划的高覆盖率序列数据上应用 Somalier 还可以识别出几个相关的样本。我们还证明它可以区分来自同一个体的全基因组和 RNA-seq 样本。
Somalier 是一种可以快速评估测序数据相关性的工具。它可以应用于不同的测序数据类型和基因组构建,并且可以在 MIT 许可证下在 github.com/brentp/somalier 上获得。