Zhang Sheng, Wang Bo, Wan Lin, Li Lei M
National Center of Mathematics and Interdisciplinary Sciences, Academy of Mathematics and Systems Science, Chinese Academy of Sciences, Beijing, 100190, China.
University of Chinese Academy of Sciences, Beijing, 100049, China.
BMC Bioinformatics. 2017 Jul 11;18(1):335. doi: 10.1186/s12859-017-1743-4.
Phred quality scores are essential for downstream DNA analysis such as SNP detection and DNA assembly. Thus a valid model to define them is indispensable for any base-calling software. Recently, we developed the base-caller 3Dec for Illumina sequencing platforms, which reduces base-calling errors by 44-69% compared to the existing ones. However, the model to predict its quality scores has not been fully investigated yet.
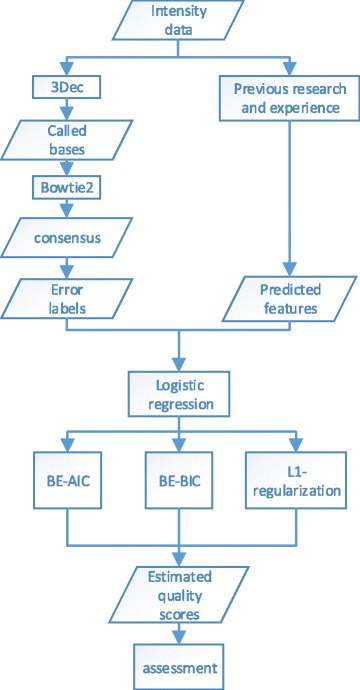
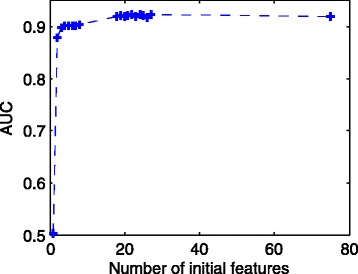
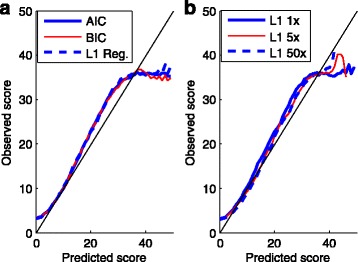
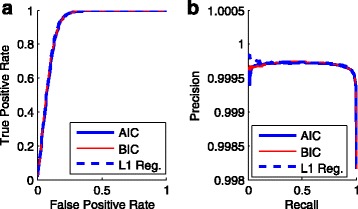
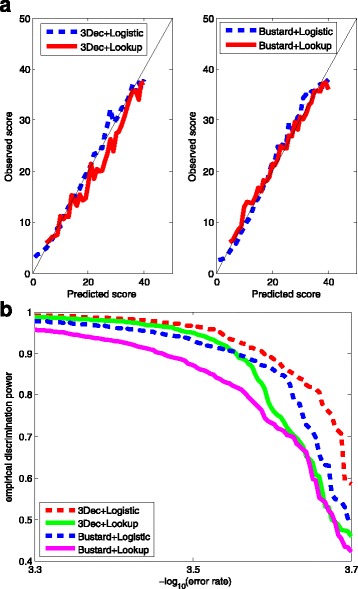
In this study, we used logistic regression models to evaluate quality scores from predictive features, which include different aspects of the sequencing signals as well as local DNA contents. Sparse models were further obtained by three methods: the backward deletion with either AIC or BIC and the L regularization learning method. The L -regularized one was then compared with the Illumina scoring method.
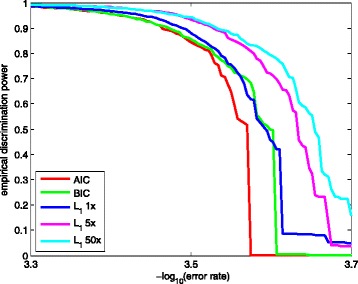
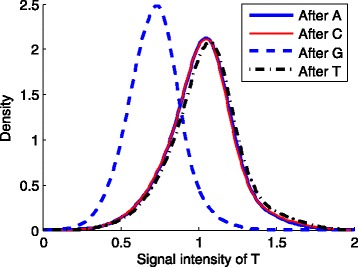
The L -regularized logistic regression improves the empirical discrimination power by as large as 14 and 25% respectively for two kinds of preprocessed sequencing signals, compared to the Illumina scoring method. Namely, the L method identifies more base calls of high fidelity. Computationally, the L method can handle large dataset and is efficient enough for daily sequencing. Meanwhile, the logistic model resulted from BIC is more interpretable. The modeling suggested that the most prominent quenching pattern in the current chemistry of Illumina occurred at the dinucleotide "GT". Besides, nucleotides were more likely to be miscalled as the previous bases if the preceding ones were not "G". It suggested that the phasing effect of bases after "G" was somewhat different from those after other nucleotide types.
Phred质量分数对于诸如单核苷酸多态性(SNP)检测和DNA组装等下游DNA分析至关重要。因此,对于任何碱基识别软件而言,一个定义这些质量分数的有效模型都是不可或缺的。最近,我们为Illumina测序平台开发了碱基识别软件3Dec,与现有软件相比,它可将碱基识别错误降低44%至69%。然而,预测其质量分数的模型尚未得到充分研究。
在本研究中,我们使用逻辑回归模型根据预测特征评估质量分数,这些预测特征包括测序信号的不同方面以及局部DNA含量。通过三种方法进一步获得了稀疏模型:使用AIC或BIC的向后删除法以及L正则化学习方法。然后将L正则化模型与Illumina评分方法进行比较。
与Illumina评分方法相比,L正则化逻辑回归分别将两种预处理测序信号的经验判别能力提高了14%和25%。也就是说,L方法识别出更多高保真度的碱基识别结果。在计算方面,L方法可以处理大型数据集,并且对于日常测序来说效率足够高。同时,由BIC得到的逻辑模型更具可解释性。该建模表明,Illumina当前化学过程中最显著的淬灭模式出现在二核苷酸“GT”处。此外,如果前一个碱基不是“G”,则核苷酸被误判为前一个碱基的可能性更大。这表明“G”之后碱基的相位效应与其他核苷酸类型之后的碱基相位效应有所不同。