Kobren Shilpa Nadimpalli, Chazelle Bernard, Singh Mona
Department of Biomedical Informatics, Harvard Medical School, Boston, MA, USA; Department of Computer Science, Princeton University, Princeton, NJ, USA; Lewis-Sigler Institute for Integrative Genomics, Princeton University, Princeton, NJ, USA.
Department of Computer Science, Princeton University, Princeton, NJ, USA.
Cell Syst. 2020 Jul 22;11(1):63-74.e7. doi: 10.1016/j.cels.2020.06.005. Epub 2020 Jul 14.
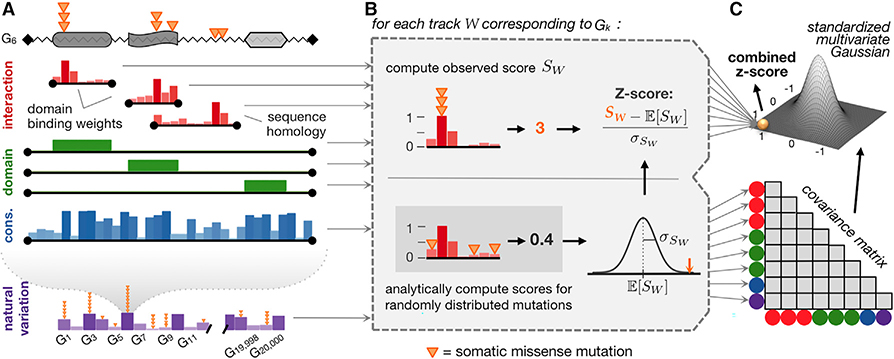
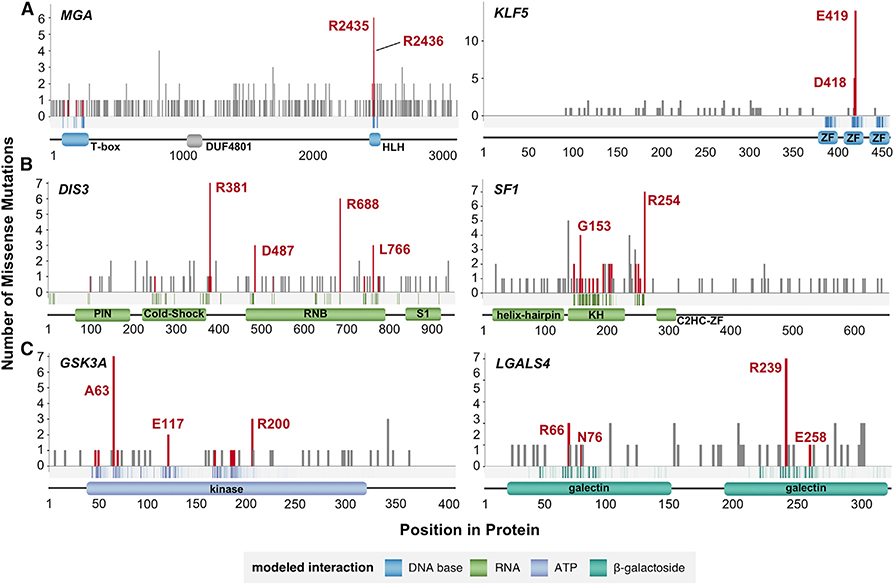
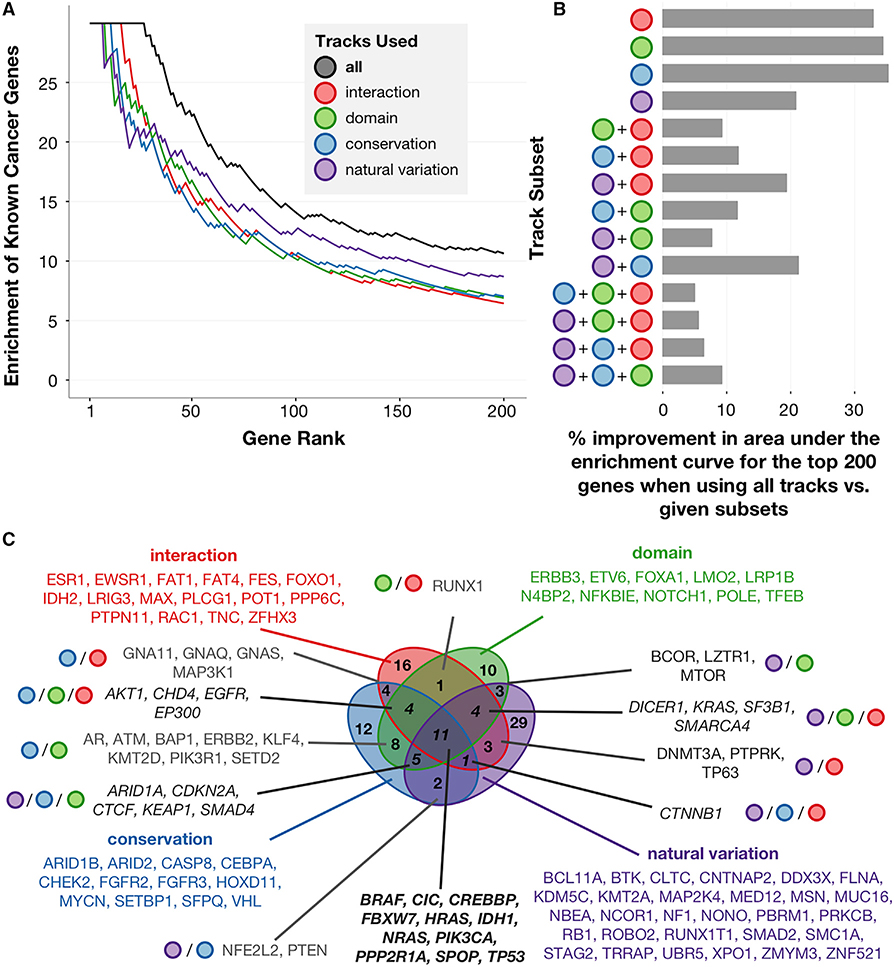
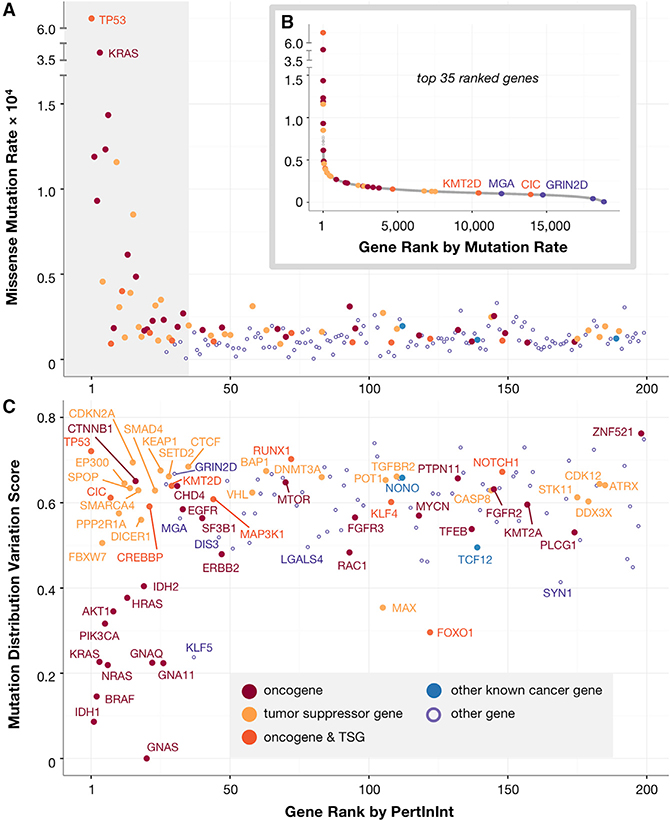
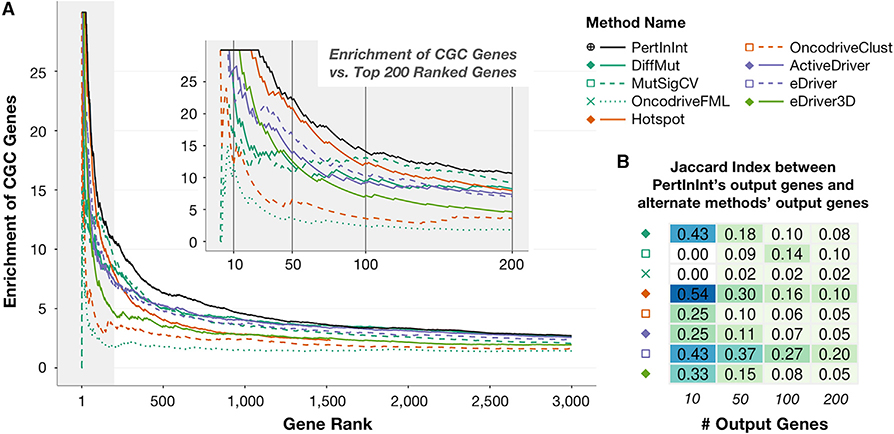
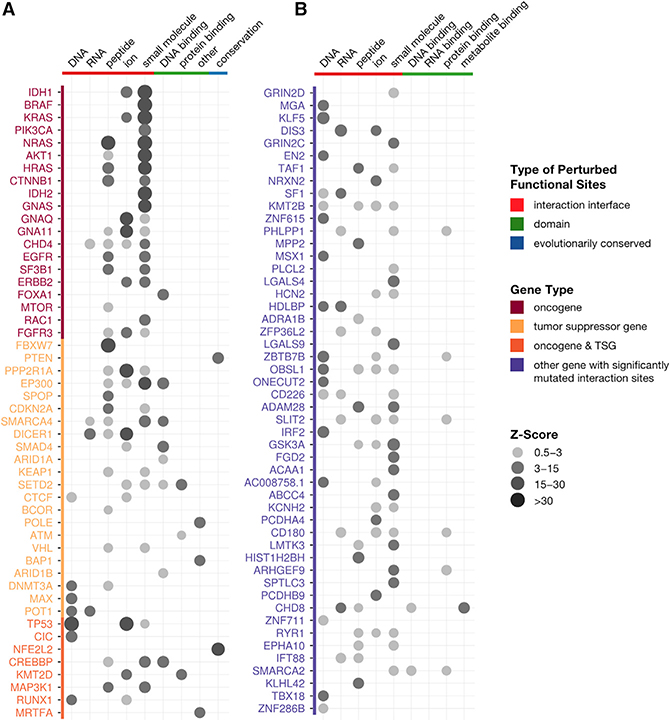
A major challenge in cancer genomics is to identify genes with functional roles in cancer and uncover their mechanisms of action. We introduce an integrative framework that identifies cancer-relevant genes by pinpointing those whose interaction or other functional sites are enriched in somatic mutations across tumors. We derive analytical calculations that enable us to avoid time-prohibitive permutation-based significance tests, making it computationally feasible to simultaneously consider multiple measures of protein site functionality. Our accompanying software, PertInInt, combines knowledge about sites participating in interactions with DNA, RNA, peptides, ions, or small molecules with domain, evolutionary conservation, and gene-level mutation data. When applied to 10,037 tumor samples, PertInInt uncovers both known and newly predicted cancer genes, while additionally revealing what types of interactions or other functionalities are disrupted. PertInInt's analysis demonstrates that somatic mutations are frequently enriched in interaction sites and domains and implicates interaction perturbation as a pervasive cancer-driving event.
癌症基因组学中的一个主要挑战是识别在癌症中具有功能作用的基因,并揭示其作用机制。我们引入了一个综合框架,通过精确确定那些其相互作用或其他功能位点在肿瘤体细胞突变中富集的基因来识别与癌症相关的基因。我们推导了分析计算方法,使我们能够避免基于排列的耗时显著检验,从而在计算上可行地同时考虑蛋白质位点功能的多种度量。我们配套的软件PertInInt,将关于参与与DNA、RNA、肽、离子或小分子相互作用的位点的知识与结构域、进化保守性和基因水平的突变数据相结合。当应用于10037个肿瘤样本时,PertInInt不仅揭示了已知的和新预测的癌症基因,还揭示了哪些类型的相互作用或其他功能被破坏。PertInInt的分析表明,体细胞突变经常在相互作用位点和结构域中富集,并表明相互作用扰动是一种普遍的癌症驱动事件。