Laboratory of DNA Damage Signaling, Department of Late Effects Studies, Radiation Biology Center, Graduate School of Biostudies, and.
Department of Hematology and Oncology, Graduate School of Medicine, Kyoto University, Kyoto, Japan.
Blood. 2021 Jan 21;137(3):336-348. doi: 10.1182/blood.2019003782.
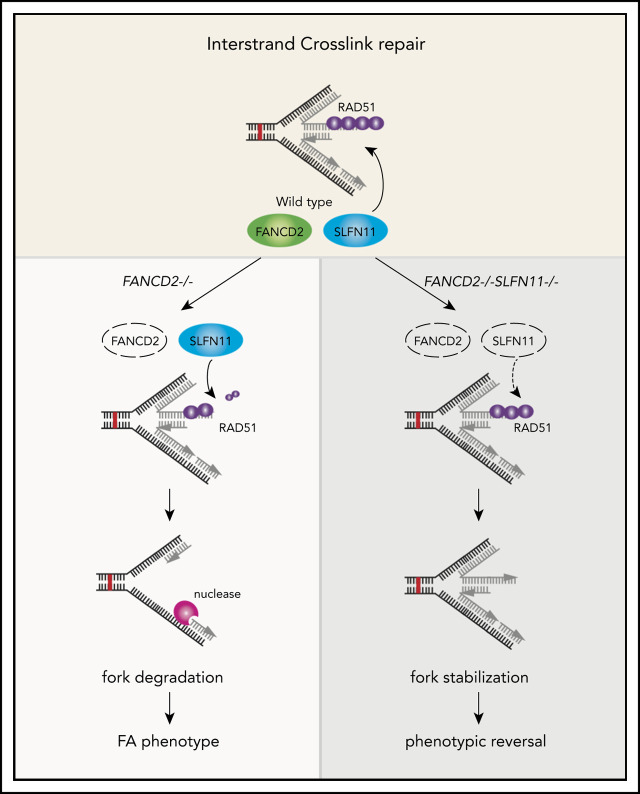
Fanconi anemia (FA) is a hereditary disorder caused by mutations in any 1 of 22 FA genes. The disease is characterized by hypersensitivity to interstrand crosslink (ICL) inducers such as mitomycin C (MMC). In addition to promoting ICL repair, FA proteins such as RAD51, BRCA2, or FANCD2 protect stalled replication forks from nucleolytic degradation during replication stress, which may have a profound impact on FA pathophysiology. Recent studies showed that expression of the putative DNA/RNA helicase SLFN11 in cancer cells correlates with cell death on chemotherapeutic treatment. However, the underlying mechanisms of SLFN11-mediated DNA damage sensitivity remain unclear. Because SLFN11 expression is high in hematopoietic stem cells, we hypothesized that SLFN11 depletion might ameliorate the phenotypes of FA cells. Here we report that SLFN11 knockdown in the FA patient-derived FANCD2-deficient PD20 cell line improved cell survival on treatment with ICL inducers. FANCD2-/-SLFN11-/- HAP1 cells also displayed phenotypic rescue, including reduced levels of MMC-induced chromosome breakage compared with FANCD2-/- cells. Importantly, we found that SLFN11 promotes extensive fork degradation in FANCD2-/- cells. The degradation process is mediated by the nucleases MRE11 or DNA2 and depends on the SLFN11 ATPase activity. This observation was accompanied by an increased RAD51 binding at stalled forks, consistent with the role of RAD51 antagonizing nuclease recruitment and subsequent fork degradation. Suppression of SLFN11 protects nascent DNA tracts even in wild-type cells. We conclude that SLFN11 destabilizes stalled replication forks, and this function may contribute to the attrition of hematopoietic stem cells in FA.
范可尼贫血症(FA)是一种由 22 个 FA 基因中的任何一个突变引起的遗传性疾病。该疾病的特征是对丝裂霉素 C(MMC)等链间交联(ICL)诱导剂高度敏感。除了促进 ICL 修复外,FA 蛋白,如 RAD51、BRCA2 或 FANCD2,在复制应激期间保护停滞的复制叉免受核酶降解,这可能对 FA 的病理生理学产生深远影响。最近的研究表明,在癌细胞中假定的 DNA/RNA 解旋酶 SLFN11 的表达与化疗治疗时的细胞死亡相关。然而,SLFN11 介导的 DNA 损伤敏感性的潜在机制尚不清楚。由于 SLFN11 在造血干细胞中的表达水平较高,我们假设 SLFN11 耗竭可能改善 FA 细胞的表型。在这里,我们报告在 FA 患者来源的 FANCD2 缺陷型 PD20 细胞系中 SLFN11 敲低可改善 ICL 诱导剂处理后的细胞存活。FANCD2-/-SLFN11-/-HAP1 细胞也显示出表型挽救,包括与 FANCD2-/-细胞相比,MMC 诱导的染色体断裂水平降低。重要的是,我们发现 SLFN11 促进 FANCD2-/-细胞中广泛的叉降解。降解过程由核酶 MRE11 或 DNA2 介导,并依赖于 SLFN11 的 ATP 酶活性。这一观察结果伴随着 RAD51 在停滞叉上的结合增加,这与 RAD51 拮抗核酶募集和随后的叉降解的作用一致。SLFN11 的抑制甚至在野生型细胞中也能保护新生 DNA 链。我们得出结论,SLFN11 使停滞的复制叉不稳定,并且该功能可能导致 FA 中的造血干细胞耗竭。