Beijing Advanced Innovation Center for Materials Genome Engineering, School of Computer and Communication Engineering, University of Science and Technology Beijing, Beijing, China.
Computational Biology Department, Carnegie Mellon University, Pittsburgh, PA, United States.
BMC Bioinformatics. 2020 Sep 9;21(1):399. doi: 10.1186/s12859-020-03660-w.
Cryo-electron tomography is an important and powerful technique to explore the structure, abundance, and location of ultrastructure in a near-native state. It contains detailed information of all macromolecular complexes in a sample cell. However, due to the compact and crowded status, the missing edge effect, and low signal to noise ratio (SNR), it is extremely challenging to recover such information with existing image processing methods. Cryo-electron tomogram simulation is an effective solution to test and optimize the performance of the above image processing methods. The simulated images could be regarded as the labeled data which covers a wide range of macromolecular complexes and ultrastructure. To approximate the crowded cellular environment, it is very important to pack these heterogeneous structures as tightly as possible. Besides, simulating non-deformable and deformable components under a unified framework also need to be achieved.
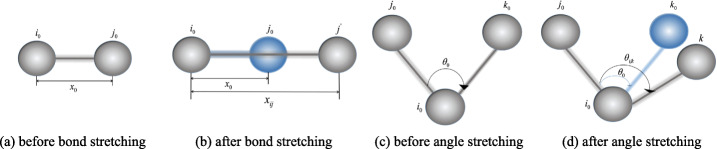
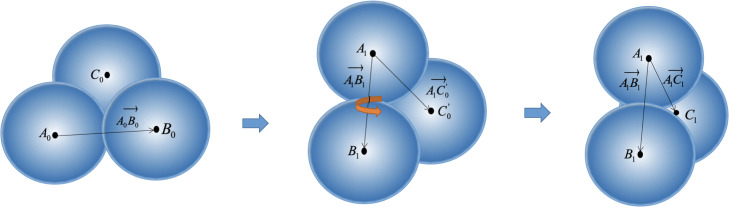
In this paper, we proposed a unified framework for simulating crowded cryo-electron tomogram images including non-deformable macromolecular complexes and deformable ultrastructures. A macromolecule was approximated using multiple balls with fixed relative positions to reduce the vacuum volume. A ultrastructure, such as membrane and filament, was approximated using multiple balls with flexible relative positions so that this structure could deform under force field. In the experiment, 400 macromolecules of 20 representative types were packed into simulated cytoplasm by our framework, and numerical verification proved that our method has a smaller volume and higher compression ratio than the baseline single-ball model. We also packed filaments, membranes and macromolecules together, to obtain a simulated cryo-electron tomogram image with deformable structures. The simulated results are closer to the real Cryo-ET, making the analysis more difficult. The DOG particle picking method and the image segmentation method are tested on our simulation data, and the experimental results show that these methods still have much room for improvement.
The proposed multi-ball model can achieve more crowded packaging results and contains richer elements with different properties to obtain more realistic cryo-electron tomogram simulation. This enables users to simulate cryo-electron tomogram images with non-deformable macromolecular complexes and deformable ultrastructures under a unified framework. To illustrate the advantages of our framework in improving the compression ratio, we calculated the volume of simulated macromolecular under our multi-ball method and traditional single-ball method. We also performed the packing experiment of filaments and membranes to demonstrate the simulation ability of deformable structures. Our method can be used to do a benchmark by generating large labeled cryo-ET dataset and evaluating existing image processing methods. Since the content of the simulated cryo-ET is more complex and crowded compared with previous ones, it will pose a greater challenge to existing image processing methods.
冷冻电子断层扫描是一种探索近天然状态下超微结构的结构、丰度和位置的重要且强大的技术。它包含样本细胞中所有大分子复合物的详细信息。然而,由于紧凑和拥挤的状态、缺失边缘效应和低信噪比(SNR),使用现有的图像处理方法恢复这些信息极具挑战性。冷冻电子断层扫描模拟是测试和优化上述图像处理方法性能的有效解决方案。模拟图像可以被视为包含广泛的大分子复合物和超微结构的标记数据。为了近似拥挤的细胞环境,尽可能紧密地包装这些异质结构非常重要。此外,还需要在统一框架下模拟不可变形和可变形组件。
本文提出了一种用于模拟包括不可变形大分子复合物和可变形超微结构的拥挤冷冻电子断层扫描图像的统一框架。使用多个具有固定相对位置的球体来近似大分子以减少真空体积。使用多个具有灵活相对位置的球体来近似膜和细丝等超微结构,以便该结构可以在力场下变形。在实验中,我们使用该框架将 400 个 20 种代表性类型的大分子包装到模拟细胞质中,数值验证证明我们的方法比基线单球模型具有更小的体积和更高的压缩比。我们还将细丝、膜和大分子一起包装,以获得具有可变形结构的模拟冷冻电子断层扫描图像。模拟结果更接近真实的冷冻电子断层扫描,使分析更加困难。我们在模拟数据上测试了 DOG 粒子选择方法和图像分割方法,实验结果表明这些方法仍有很大的改进空间。
所提出的多球模型可以实现更拥挤的包装结果,并包含更多具有不同性质的元素,以获得更真实的冷冻电子断层扫描模拟。这使我们能够在统一框架下模拟具有不可变形大分子复合物和可变形超微结构的冷冻电子断层扫描图像。为了说明我们的框架在提高压缩比方面的优势,我们计算了使用我们的多球方法和传统的单球方法模拟的大分子的体积。我们还进行了细丝和膜的包装实验,以证明可变形结构的模拟能力。我们的方法可以通过生成大型标记冷冻电子断层扫描数据集并评估现有的图像处理方法来用作基准。由于模拟的冷冻电子断层扫描内容比以前的内容更复杂和拥挤,因此现有的图像处理方法将面临更大的挑战。