CINBIO, Universidade de Vigo, Vigo, Spain.
Instituto de Investigación Sanitaria Galicia Sur, Hospital Álvaro Cunqueiro, Vigo, Spain.
Sci Rep. 2020 Sep 15;10(1):15135. doi: 10.1038/s41598-020-72089-1.
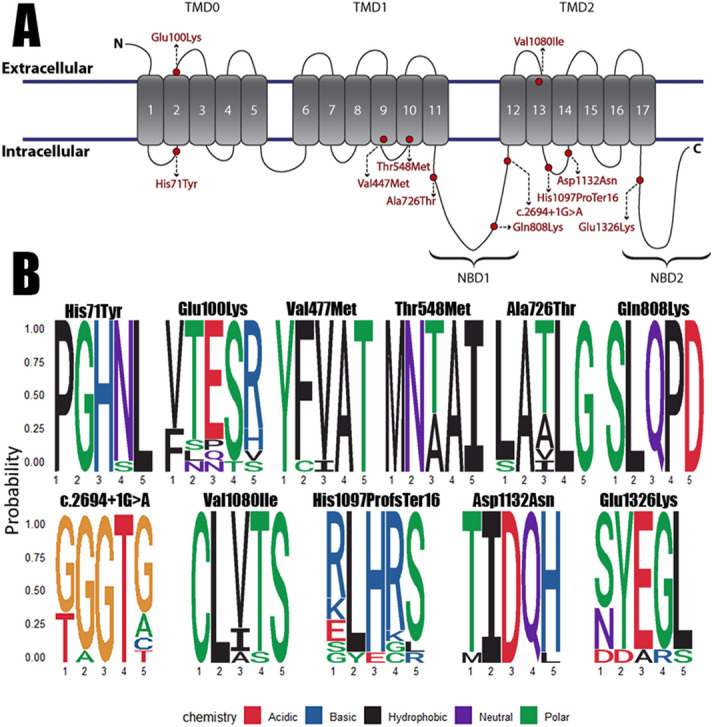
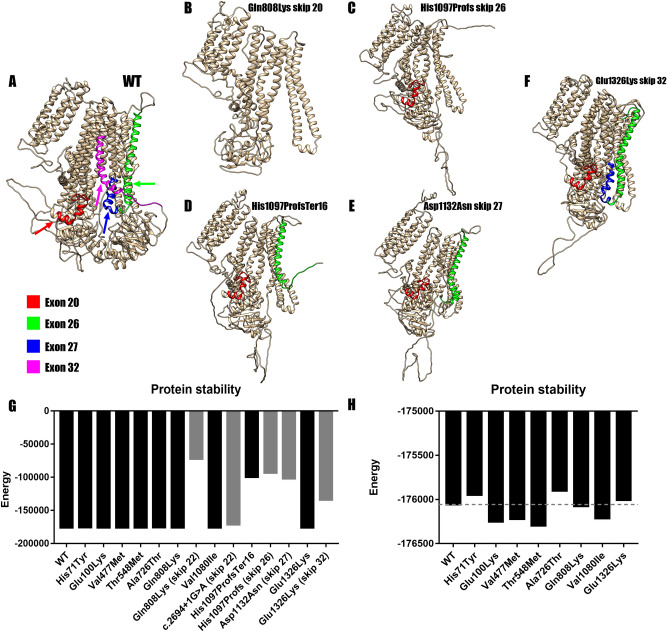
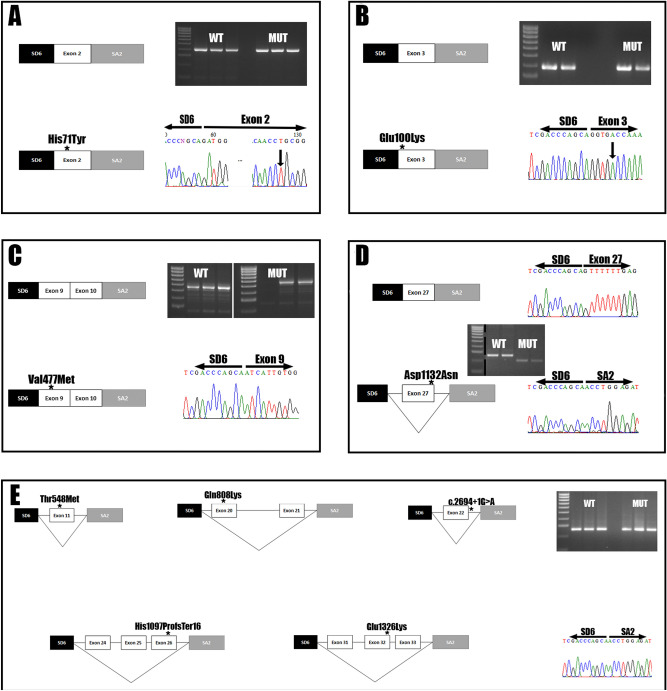
Pulmonary Arterial Hypertension (PAH) is a rare and fatal disease where knowledge about its genetic basis continues to increase. In this study, we used targeted panel sequencing in a cohort of 624 adult and pediatric patients from the Spanish PAH registry. We identified 11 rare variants in the ATP-binding Cassette subfamily C member 8 (ABCC8) gene, most of them with splicing alteration predictions. One patient also carried another variant in SMAD1 gene (c.27delinsGTAAAG). We performed an ABCC8 in vitro biochemical analyses using hybrid minigenes to confirm the correct mRNA processing of 3 missense variants (c.211C > T p.His71Tyr, c.298G > A p.Glu100Lys and c.1429G > A p.Val477Met) and the skipping of exon 27 in the novel splicing variant c.3394G > A. Finally, we used structural protein information to further assess the pathogenicity of the variants. The results showed 11 novel changes in ABCC8 and 1 in SMAD1 present in PAH patients. After in silico and in vitro biochemical analyses, we classified 2 as pathogenic (c.3288_3289del and c.3394G > A), 6 as likely pathogenic (c.211C > T, c.1429G > A, c.1643C > T, c.2422C > A, c.2694 + 1G > A, c.3976G > A and SMAD1 c.27delinsGTAAAG) and 3 as Variants of Uncertain Significance (c.298G > A, c.2176G > A and c.3238G > A). In all, we show that coupling in silico tools with in vitro biochemical studies can improve the classification of genetic variants.
肺动脉高压(PAH)是一种罕见且致命的疾病,其遗传基础的相关知识不断增加。在这项研究中,我们使用靶向panel 测序对来自西班牙 PAH 注册中心的 624 名成年和儿科患者进行了研究。我们在 ATP 结合盒亚家族 C 成员 8(ABCC8)基因中发现了 11 个罕见变异,其中大多数具有剪接改变的预测。一名患者还携带 SMAD1 基因(c.27delinsGTAAAG)的另一个变异。我们使用杂交 minigene 对 ABCC8 进行了体外生化分析,以确认 3 个错义变异(c.211C>T p.His71Tyr、c.298G>A p.Glu100Lys 和 c.1429G>A p.Val477Met)和外显子 27 缺失的正确 mRNA 处理,并在新的剪接变异 c.3394G>A 中发现了剪接改变。最后,我们使用结构蛋白信息进一步评估变异的致病性。结果显示,在 PAH 患者中发现了 ABCC8 的 11 个新变化和 SMAD1 的 1 个变化。经过计算机预测和体外生化分析,我们将 2 个变异归类为致病性(c.3288_3289del 和 c.3394G>A),6 个变异归类为可能致病性(c.211C>T、c.1429G>A、c.1643C>T、c.2422C>A、c.2694+1G>A、c.3976G>A 和 SMAD1 c.27delinsGTAAAG),3 个变异归类为意义不明的变异(c.298G>A、c.2176G>A 和 c.3238G>A)。总之,我们表明,将计算机预测工具与体外生化研究相结合,可以提高遗传变异的分类。