Chen Jieming, Madireddi Shravan, Nagarkar Deepti, Migdal Maciej, Vander Heiden Jason, Chang Diana, Mukhyala Kiran, Selvaraj Suresh, Kadel Edward E, Brauer Matthew J, Mariathasan Sanjeev, Hunkapiller Julie, Jhunjhunwala Suchit, Albert Matthew L, Hammer Christian
Department of Bioinformatics and Computational Biology.
Department of Cancer Immunology.
Brief Bioinform. 2021 May 20;22(3). doi: 10.1093/bib/bbaa223.
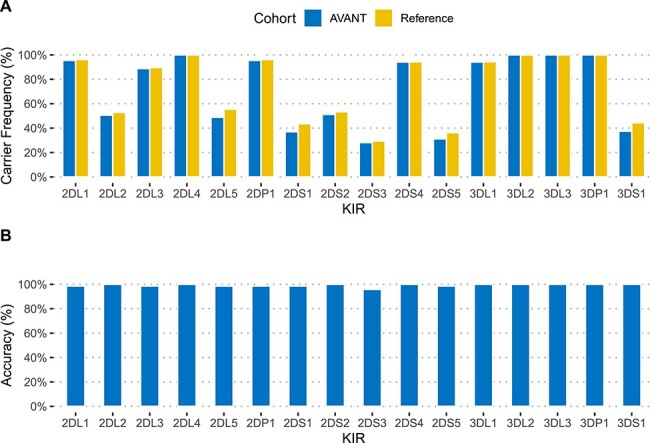
Immunogenetic variation in humans is important in research, clinical diagnosis and increasingly a target for therapeutic intervention. Two highly polymorphic loci play critical roles, namely the human leukocyte antigen (HLA) system, which is the human version of the major histocompatibility complex (MHC), and the Killer-cell immunoglobulin-like receptors (KIR) that are relevant for responses of natural killer (NK) and some subsets of T cells. Their accurate classification has typically required the use of dedicated biological specimens and a combination of in vitro and in silico efforts. Increased availability of next generation sequencing data has led to the development of ancillary computational solutions. Here, we report an evaluation of recently published algorithms to computationally infer complex immunogenetic variation in the form of HLA alleles and KIR haplotypes from whole-genome or whole-exome sequencing data. For both HLA allele and KIR gene typing, we identified tools that yielded >97% overall accuracy for four-digit HLA types, and >99% overall accuracy for KIR gene presence, suggesting the readiness of in silico solutions for use in clinical and high-throughput research settings.
人类免疫遗传学变异在研究、临床诊断中具有重要意义,并且越来越成为治疗干预的靶点。两个高度多态性位点发挥着关键作用,即人类白细胞抗原(HLA)系统,它是主要组织相容性复合体(MHC)的人类版本,以及杀伤细胞免疫球蛋白样受体(KIR),其与自然杀伤(NK)细胞和某些T细胞亚群的反应相关。它们的准确分类通常需要使用专门的生物样本以及体外和计算机模拟方法的结合。新一代测序数据可用性的提高导致了辅助计算解决方案的发展。在此,我们报告了对最近发表的算法的评估,这些算法可从全基因组或全外显子组测序数据中以HLA等位基因和KIR单倍型的形式计算推断复杂的免疫遗传学变异。对于HLA等位基因和KIR基因分型,我们鉴定出了一些工具,对于四位数字的HLA类型,其总体准确率>97%,对于KIR基因的存在情况,总体准确率>99%,这表明计算机模拟解决方案已准备好在临床和高通量研究环境中使用。