Swammerdam Institute for Life Sciences, University of Amsterdam.
PLoS Comput Biol. 2020 Sep 30;16(9):e1008295. doi: 10.1371/journal.pcbi.1008295. eCollection 2020 Sep.
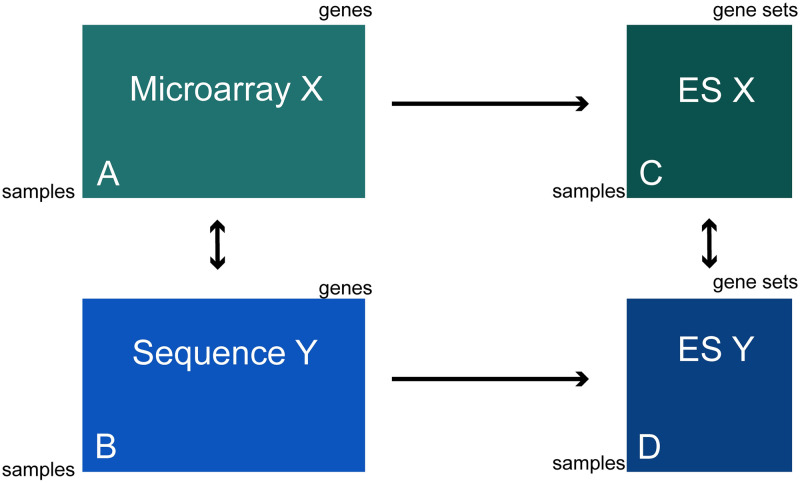
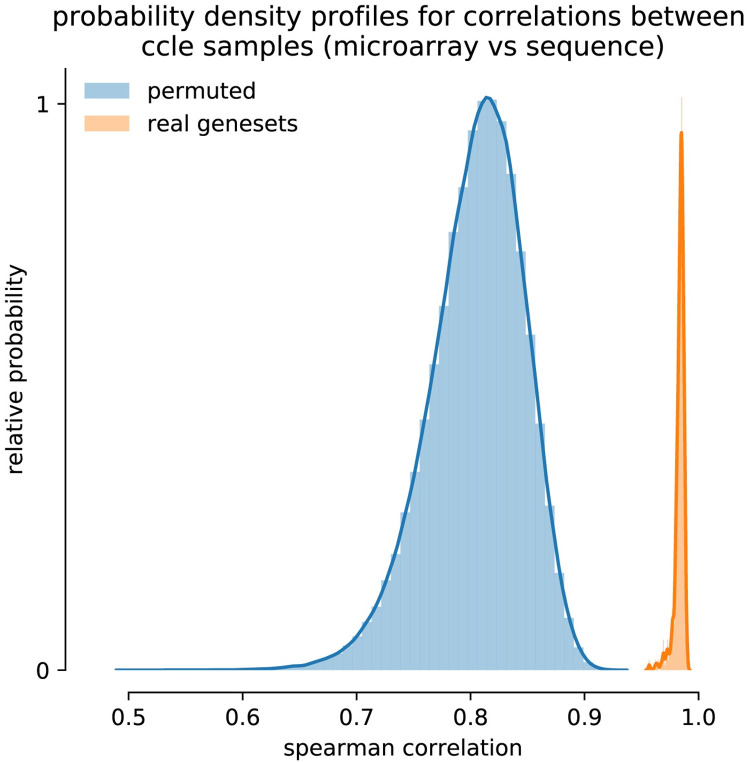
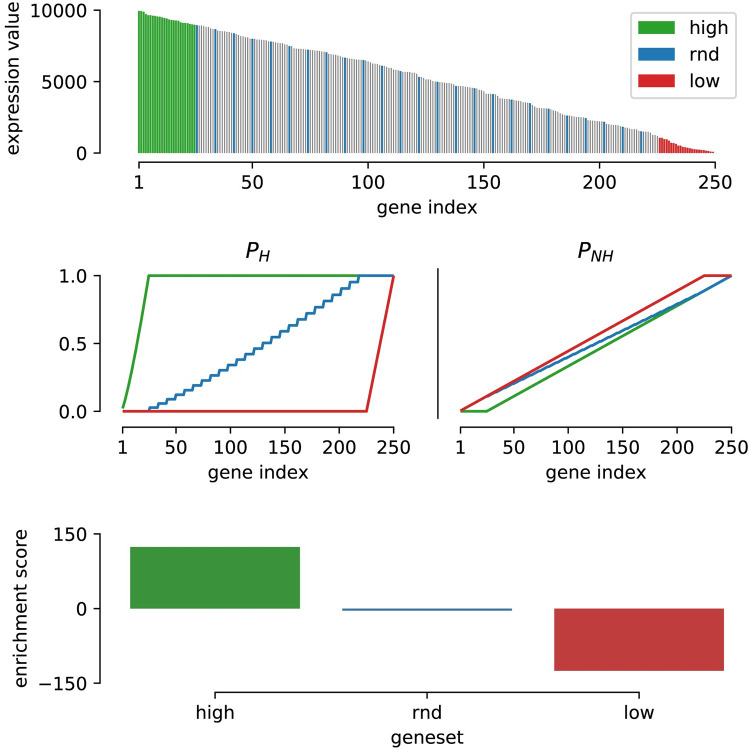
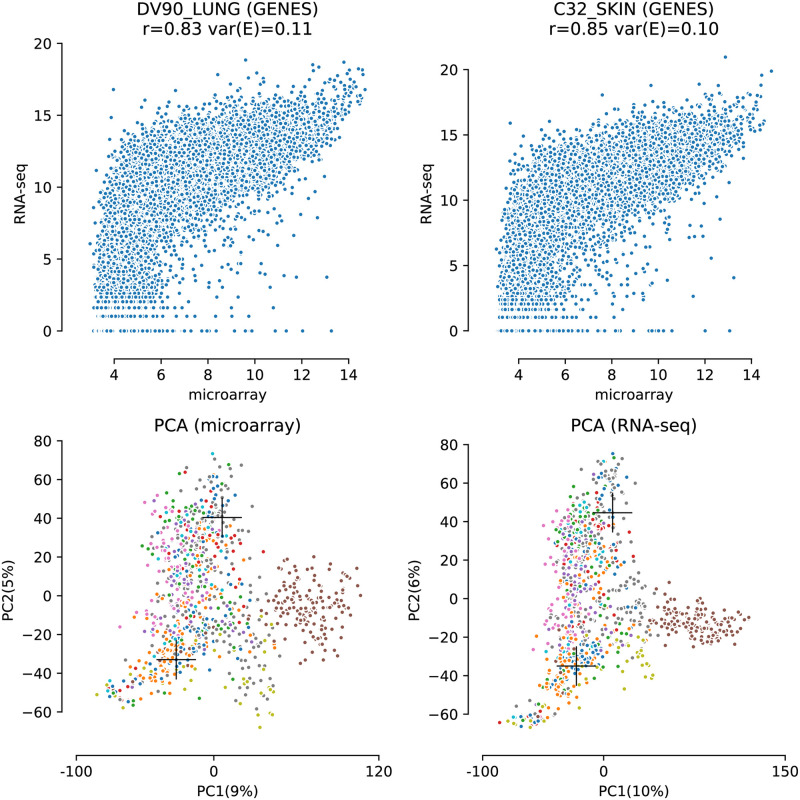
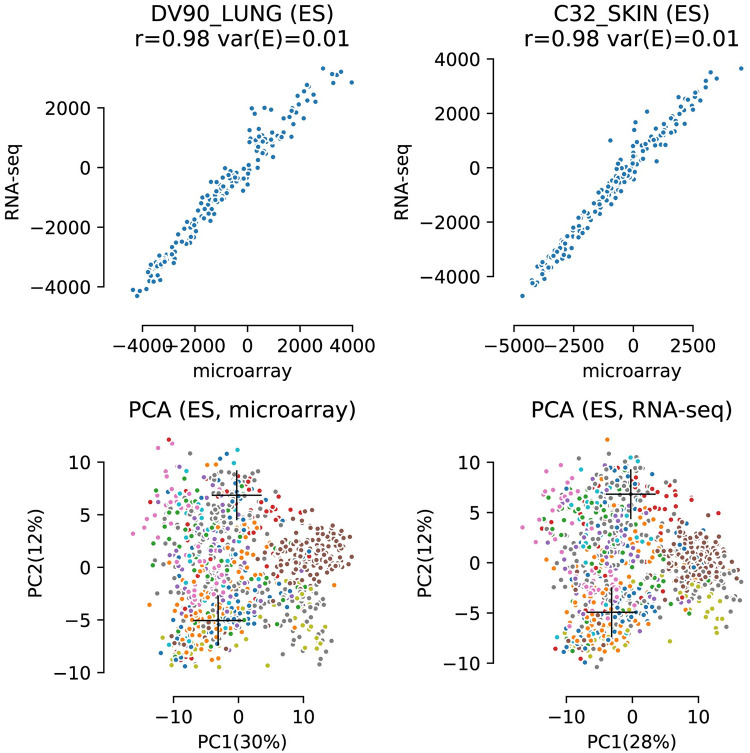
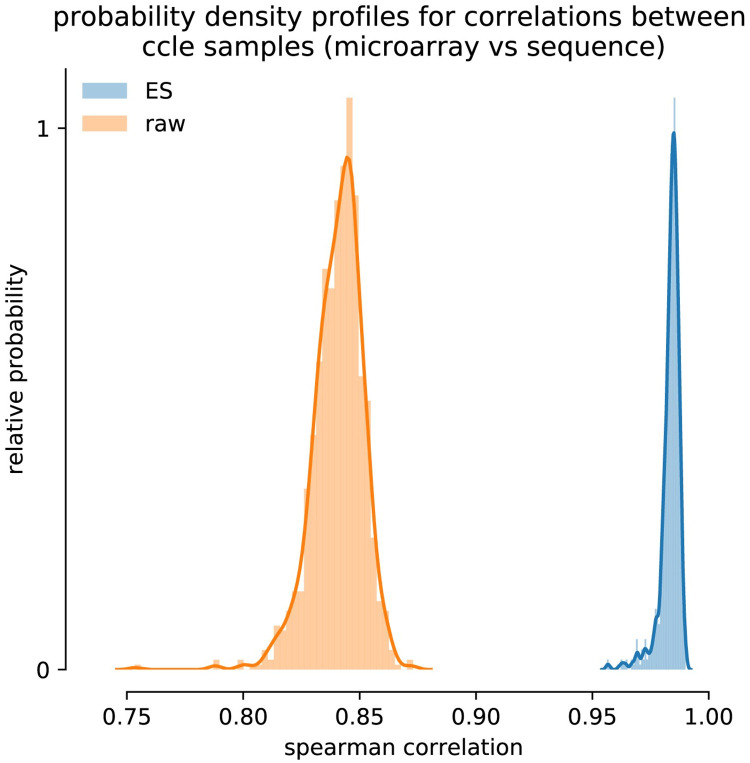
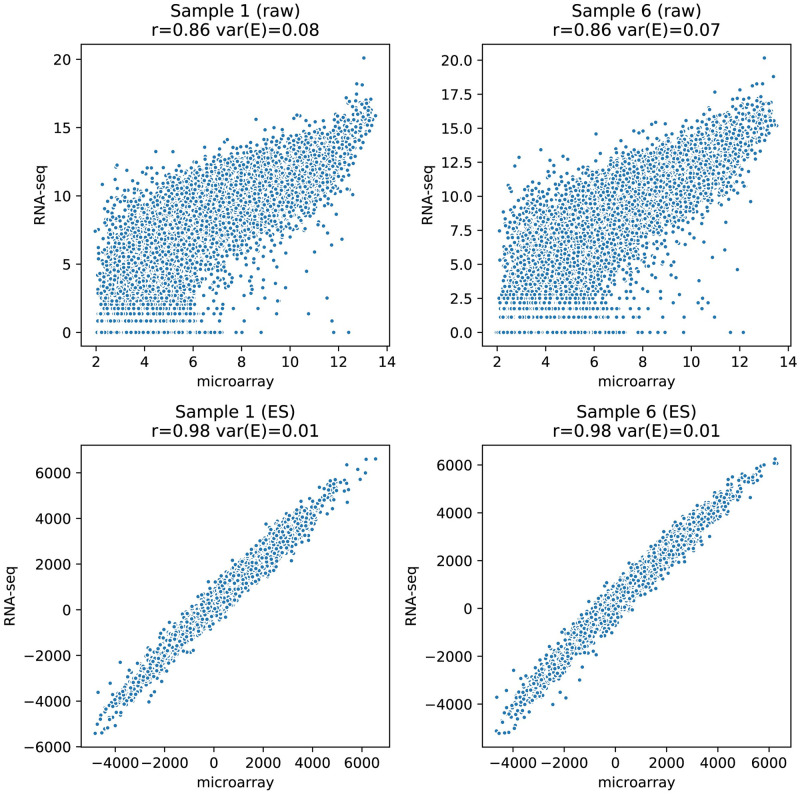
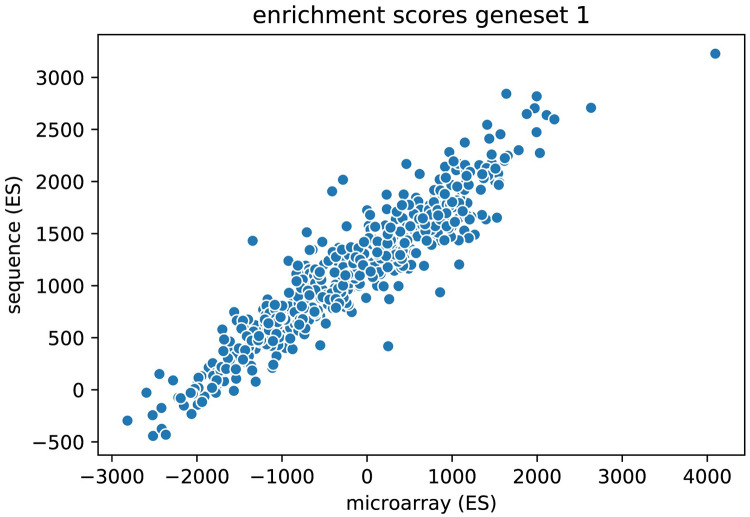
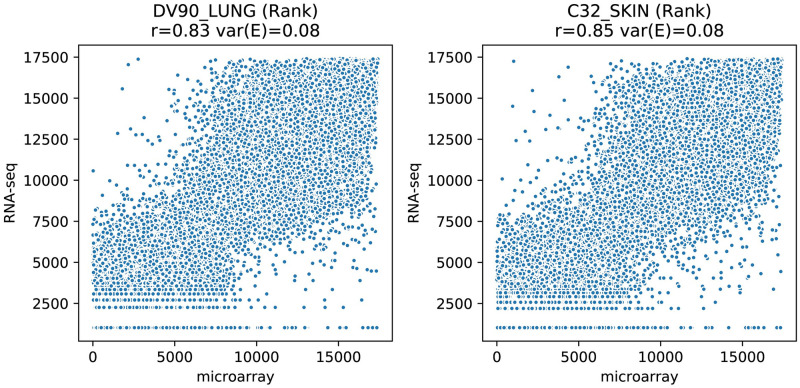
The field of transcriptomics uses and measures mRNA as a proxy of gene expression. There are currently two major platforms in use for quantifying mRNA, microarray and RNA-Seq. Many comparative studies have shown that their results are not always consistent. In this study we aim to find a robust method to increase comparability of both platforms enabling data analysis of merged data from both platforms. We transformed high dimensional transcriptomics data from two different platforms into a lower dimensional, and biologically relevant dataset by calculating enrichment scores based on gene set collections for all samples. We compared the similarity between data from both platforms based on the raw data and on the enrichment scores. We show that the performed data transforms the data in a biologically relevant way and filters out noise which leads to increased platform concordance. We validate the procedure using predictive models built with microarray based enrichment scores to predict subtypes of breast cancer using enrichment scores based on sequenced data. Although microarray and RNA-Seq expression levels might appear different, transforming them into biologically relevant gene set enrichment scores significantly increases their correlation, which is a step forward in data integration of the two platforms. The gene set collections were shown to contain biologically relevant gene sets. More in-depth investigation on the effect of the composition, size, and number of gene sets that are used for the transformation is suggested for future research.
转录组学领域使用和测量 mRNA 作为基因表达的替代物。目前有两种主要的平台用于定量 mRNA,即微阵列和 RNA-Seq。许多比较研究表明,它们的结果并不总是一致的。在这项研究中,我们旨在找到一种稳健的方法来提高这两个平台的可比性,从而能够对来自这两个平台的合并数据进行数据分析。我们通过为所有样本的基因集集合计算富集分数,将来自两个不同平台的高维转录组学数据转换为低维的、具有生物学意义的数据集。我们基于原始数据和富集分数比较了两个平台的数据之间的相似性。我们表明,所进行的数据转换以生物学上相关的方式对数据进行了转换,并过滤掉了导致平台一致性增加的噪声。我们使用基于微阵列的富集分数构建的预测模型来验证该过程,该模型使用基于测序数据的富集分数来预测乳腺癌亚型。虽然微阵列和 RNA-Seq 的表达水平可能看起来不同,但将它们转化为具有生物学意义的基因集富集分数可以显著提高它们的相关性,这是整合这两个平台数据的一个重要步骤。基因集集合被证明包含具有生物学意义的基因集。建议未来的研究更深入地研究用于转换的基因集的组成、大小和数量对其效果的影响。