North Dakota State University, 1340 Administration Ave, Fargo, 58102, USA.
BMC Bioinformatics. 2020 Sep 30;21(Suppl 14):369. doi: 10.1186/s12859-020-03689-x.
Chromosome conformation capture-based methods, especially Hi-C, enable scientists to detect genome-wide chromatin interactions and study the spatial organization of chromatin, which plays important roles in gene expression regulation, DNA replication and repair etc. Thus, developing computational methods to unravel patterns behind the data becomes critical. Existing computational methods focus on intrachromosomal interactions and ignore interchromosomal interactions partly because there is no prior knowledge for interchromosomal interactions and the frequency of interchromosomal interactions is much lower while the search space is much larger. With the development of single-cell technologies, the advent of single-cell Hi-C makes interrogating the spatial structure of chromatin at single-cell resolution possible. It also brings a new type of frequency information, the number of single cells with chromatin interactions between two disjoint chromosome regions.
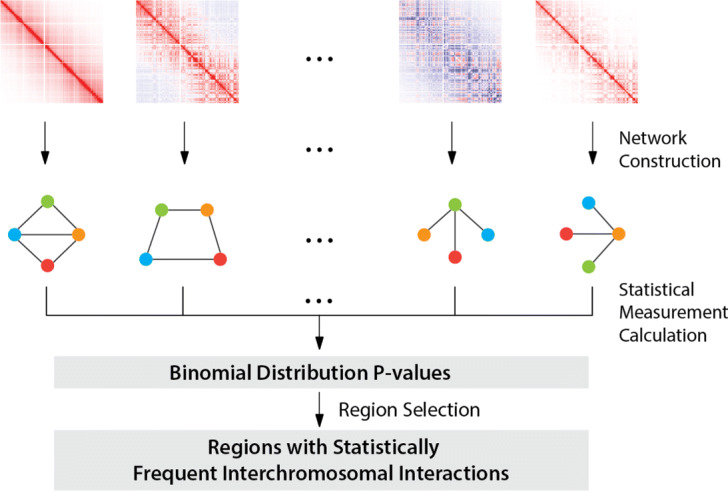
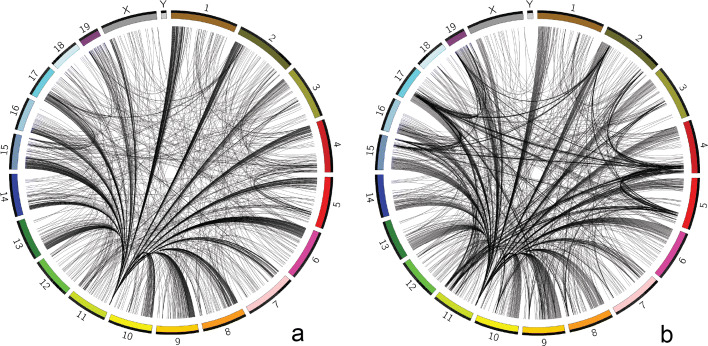
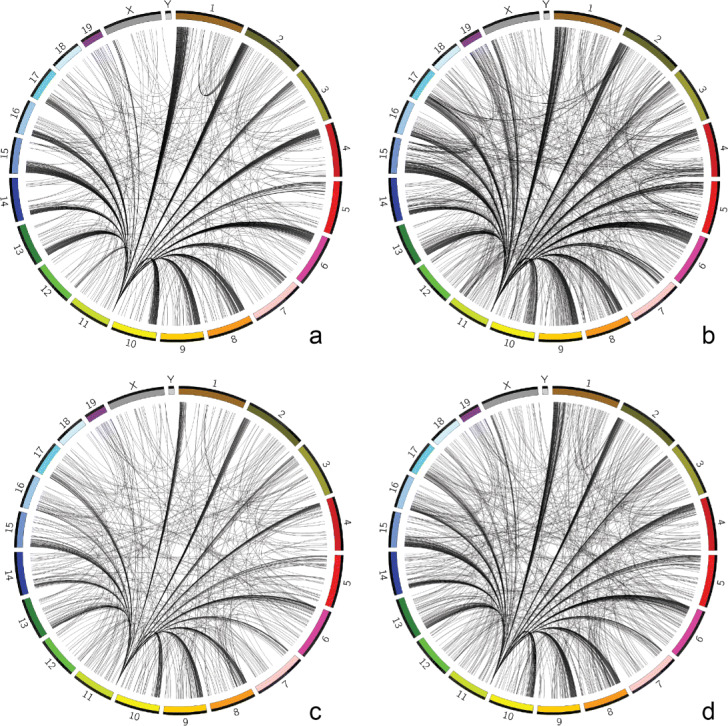
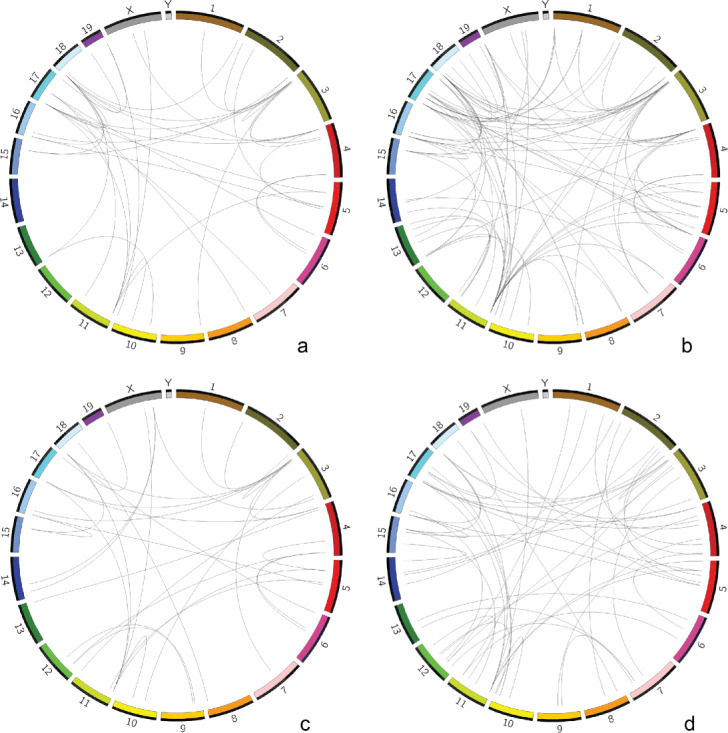
Considering the lack of computational methods on interchromosomal interactions and the unsurprisingly frequent intrachromosomal interactions along the diagonal of a chromatin contact map, we propose a computational method dedicated to analyzing interchromosomal interactions of single-cell Hi-C with this new frequency information. To the best of our knowledge, our proposed tool is the first to identify regions with statistically frequent interchromosomal interactions at single-cell resolution. We demonstrate that the tool utilizing networks and binomial statistical tests can identify interesting structural regions through visualization, comparison and enrichment analysis and it also supports different configurations to provide users with flexibility.
It will be a useful tool for analyzing single-cell Hi-C interchromosomal interactions.
基于染色体构象捕获的方法,特别是 Hi-C,可以使科学家检测全基因组染色质相互作用,并研究染色质的空间组织,这在基因表达调控、DNA 复制和修复等方面发挥着重要作用。因此,开发用于揭示数据背后模式的计算方法变得至关重要。现有的计算方法主要关注染色体内部的相互作用,而部分忽略了染色体间的相互作用,部分原因是缺乏染色体间相互作用的先验知识,而且染色体间相互作用的频率要低得多,而搜索空间要大得多。随着单细胞技术的发展,单细胞 Hi-C 的出现使得在单细胞分辨率下研究染色质的空间结构成为可能。它还带来了一种新的频率信息,即两个不相交的染色体区域之间存在染色质相互作用的单细胞数量。
考虑到缺乏染色体间相互作用的计算方法,以及在染色质接触图的对角线上沿着染色体的相互作用不可避免地频繁出现,我们提出了一种专门用于分析单细胞 Hi-C 中染色体间相互作用的计算方法,并利用这种新的频率信息。据我们所知,我们提出的工具是第一个在单细胞分辨率下识别具有统计学上频繁染色体间相互作用的区域的工具。我们证明,该工具利用网络和二项式统计检验,可以通过可视化、比较和富集分析来识别有趣的结构区域,并且它还支持不同的配置,为用户提供灵活性。
它将成为分析单细胞 Hi-C 染色体间相互作用的有用工具。