Joint Carnegie Mellon University-University of Pittsburgh Ph.D. Program in Computational Biology, Pittsburgh, PA, 15213, USA.
Computational Biology Department, School of Computer Science, Carnegie Mellon University, Pittsburgh, PA, 15213, USA.
Nat Commun. 2019 Nov 7;10(1):5069. doi: 10.1038/s41467-019-12954-4.
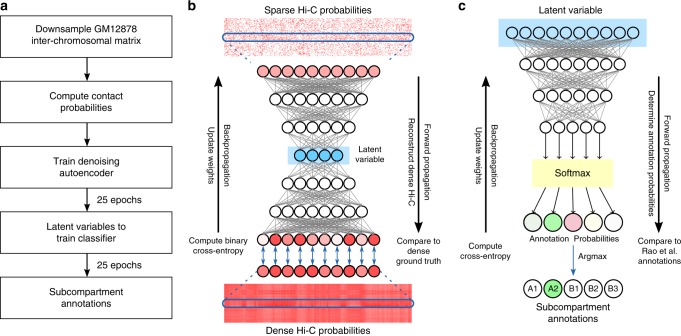
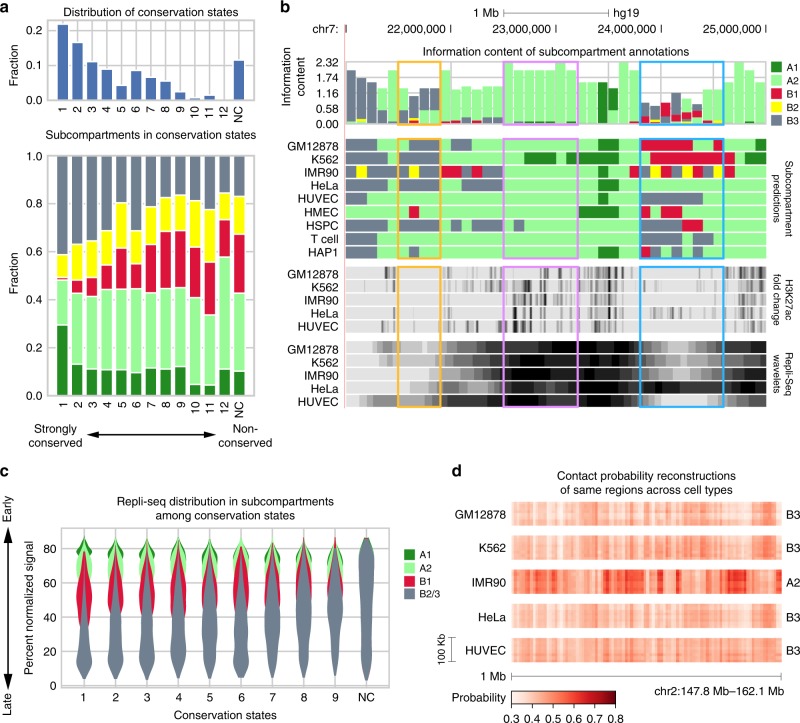
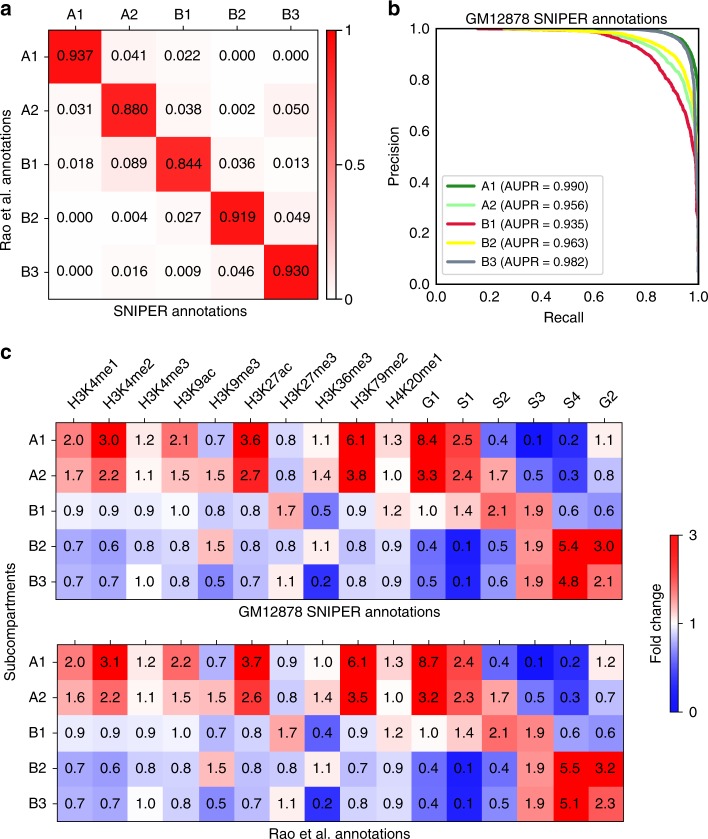
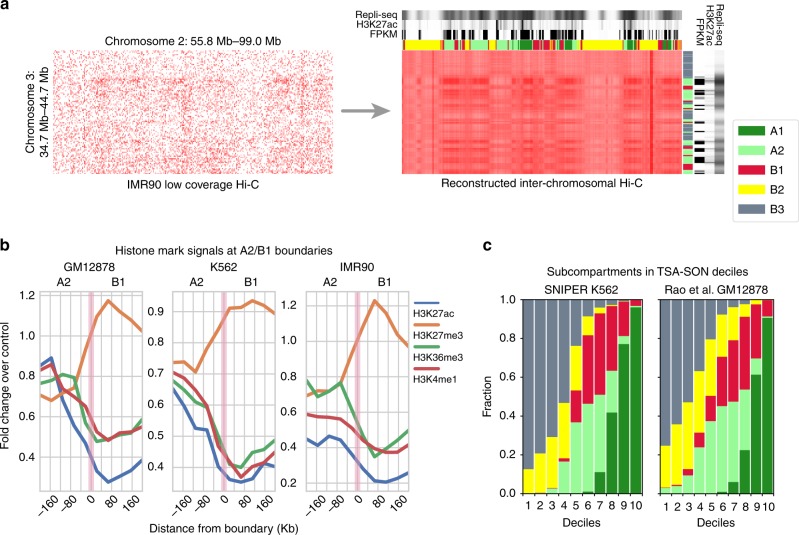
Higher-order genome organization and its variation in different cellular conditions remain poorly understood. Recent high-coverage genome-wide chromatin interaction mapping using Hi-C has revealed spatial segregation of chromosomes in the human genome into distinct subcompartments. However, subcompartment annotation, which requires Hi-C data with high sequencing coverage, is currently only available in the GM12878 cell line, making it impractical to compare subcompartment patterns across cell types. Here we develop a computational approach, SNIPER (Subcompartment iNference using Imputed Probabilistic ExpRessions), based on denoising autoencoder and multilayer perceptron classifier to infer subcompartments using typical Hi-C datasets with moderate coverage. SNIPER accurately reveals subcompartments using moderate coverage Hi-C datasets and outperforms an existing method that uses epigenomic features in GM12878. We apply SNIPER to eight additional cell lines and find that chromosomal regions with conserved and cell-type specific subcompartment annotations have different patterns of functional genomic features. SNIPER enables the identification of subcompartments without high-coverage Hi-C data and provides insights into the function and mechanisms of spatial genome organization variation across cell types.
高级基因组组织及其在不同细胞状态下的变化仍知之甚少。最近使用 Hi-C 的高覆盖率全基因组染色质互作图谱揭示了人类基因组中染色体的空间分离成不同的亚区室。然而,亚区室注释需要具有高测序覆盖率的 Hi-C 数据,目前仅在 GM12878 细胞系中可用,使得跨细胞类型比较亚区室模式变得不切实际。在这里,我们开发了一种基于去噪自动编码器和多层感知机分类器的计算方法 SNIPER(使用推断的概率表达进行亚区室推断),用于使用中等覆盖度的典型 Hi-C 数据集推断亚区室。SNIPER 使用中等覆盖度的 Hi-C 数据集准确地揭示了亚区室,并且优于使用 GM12878 中的表观遗传特征的现有方法。我们将 SNIPER 应用于另外 8 个细胞系,并发现具有保守和细胞类型特异性亚区室注释的染色体区域具有不同的功能基因组特征模式。SNIPER 能够在没有高覆盖率 Hi-C 数据的情况下识别亚区室,并深入了解跨细胞类型的空间基因组组织变化的功能和机制。