Infectious Diseases, Internal Medicine, Michigan Medicine, University of Michigan, Ann Arbor, MI, USA.
Microbiome Research Initiative, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave N, E4-100, Seattle, WA, 98109-1024, USA.
BMC Bioinformatics. 2020 Oct 15;21(1):459. doi: 10.1186/s12859-020-03802-0.
High-throughput sequencing can establish the functional capacity of a microbial community by cataloging the protein-coding sequences (CDS) present in the metagenome of the community. The relative performance of different computational methods for identifying CDS from whole-genome shotgun sequencing is not fully established.
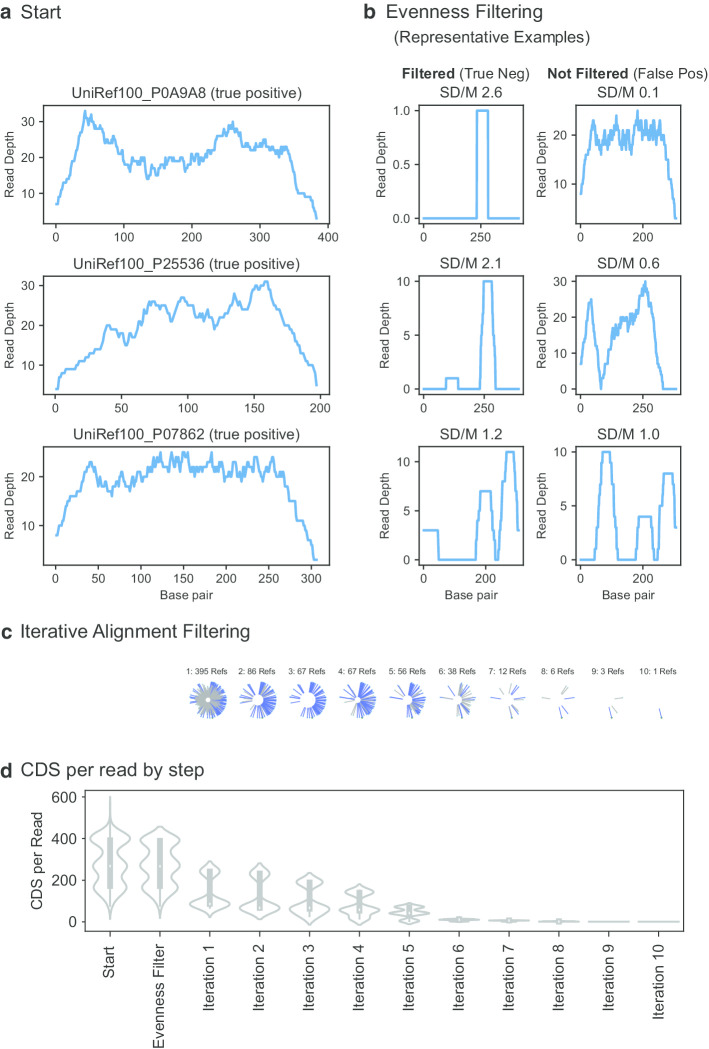
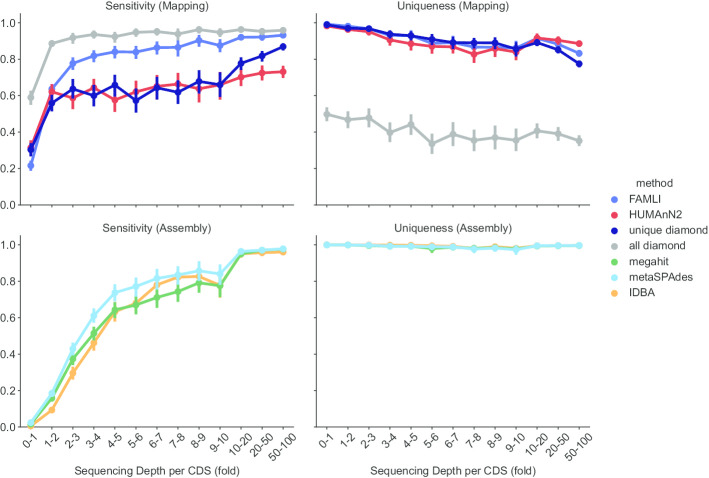
Here we present an automated benchmarking workflow, using synthetic shotgun sequencing reads for which we know the true CDS content of the underlying communities, to determine the relative performance (sensitivity, positive predictive value or PPV, and computational efficiency) of different metagenome analysis tools for extracting the CDS content of a microbial community. Assembly-based methods are limited by coverage depth, with poor sensitivity for CDS at < 5X depth of sequencing, but have excellent PPV. Mapping-based techniques are more sensitive at low coverage depths, but can struggle with PPV. We additionally describe an expectation maximization based iterative algorithmic approach which we show to successfully improve the PPV of a mapping based technique while retaining improved sensitivity and computational efficiency.
Our benchmarking approach reveals the trade-offs of assembly versus alignment-based approaches and the relative performance of specific implementations when one wishes to extract the protein coding capacity of microbial communities.
高通量测序可以通过对群落宏基因组中存在的蛋白编码序列(CDS)进行编目,从而建立微生物群落的功能能力。不同计算方法在识别全基因组鸟枪法测序中 CDS 的相对性能尚未完全确定。
在这里,我们提出了一种自动化的基准测试工作流程,使用我们知道潜在群落中真实 CDS 内容的合成鸟枪法测序reads,以确定不同宏基因组分析工具提取微生物群落 CDS 内容的相对性能(灵敏度、阳性预测值或 PPV 和计算效率)。基于组装的方法受覆盖深度限制,对于测序深度 < 5X 的 CDS 灵敏度较差,但 PPV 非常高。基于映射的技术在低覆盖深度下更敏感,但可能难以获得 PPV。我们还描述了一种基于期望最大化的迭代算法方法,我们证明该方法可以成功提高基于映射的技术的 PPV,同时保持改进的灵敏度和计算效率。
我们的基准测试方法揭示了组装与基于比对的方法之间的权衡,以及当希望提取微生物群落的蛋白质编码能力时,特定实现的相对性能。