Data Core, Fred Hutchinson Cancer Center, Seattle, WA, USA.
Bioinformatics Graduate Program, University of Michigan, Ann Arbor, MI, USA.
Cell Rep Methods. 2023 Nov 20;3(11):100639. doi: 10.1016/j.crmeth.2023.100639. Epub 2023 Nov 7.
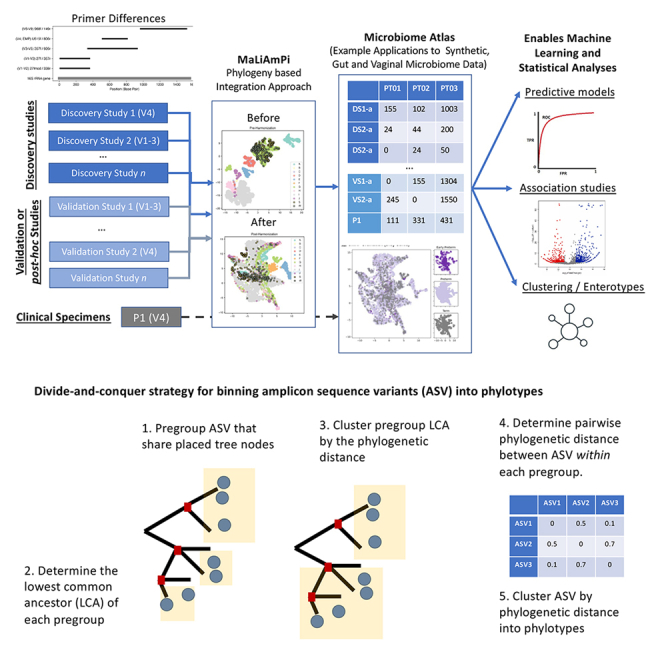
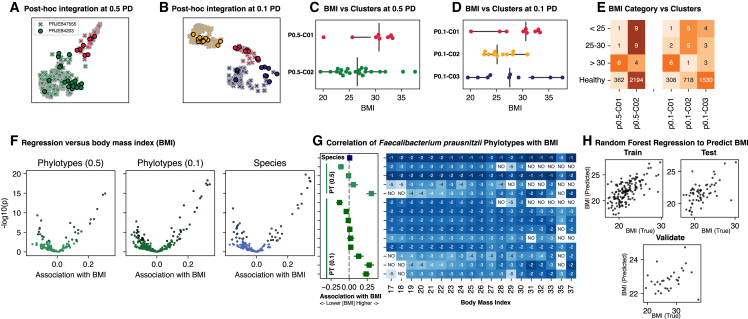
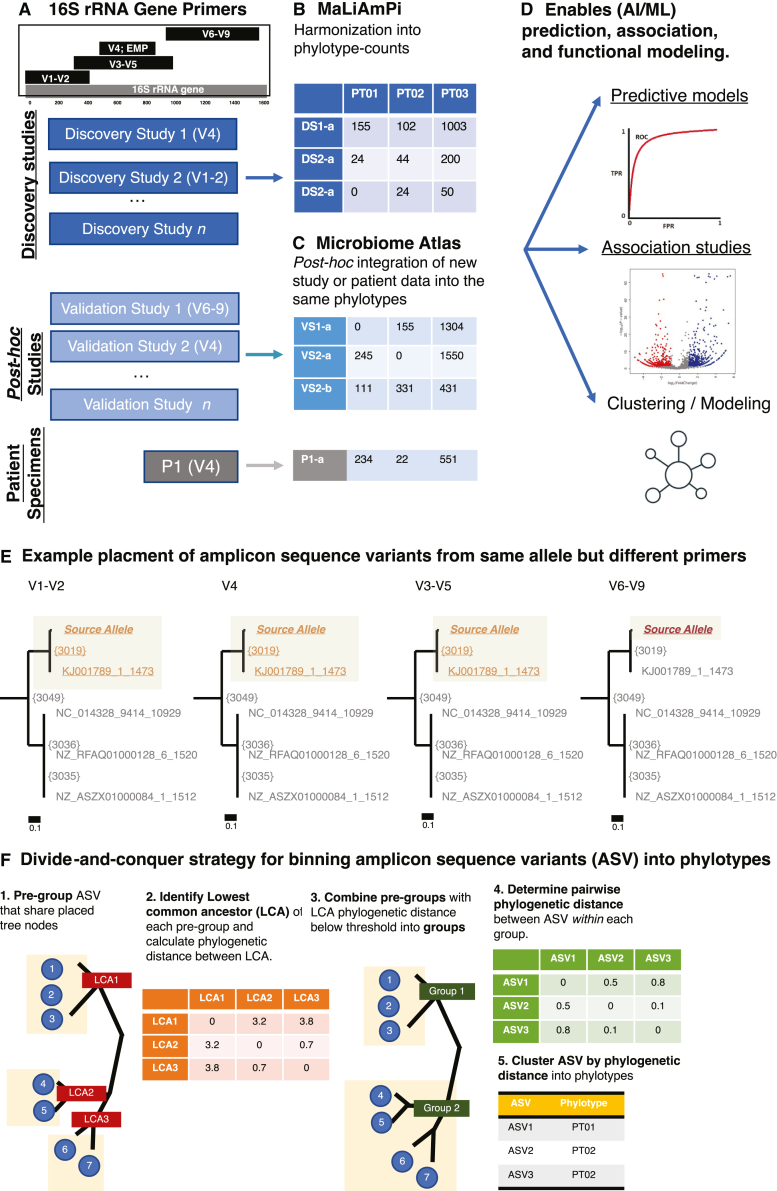
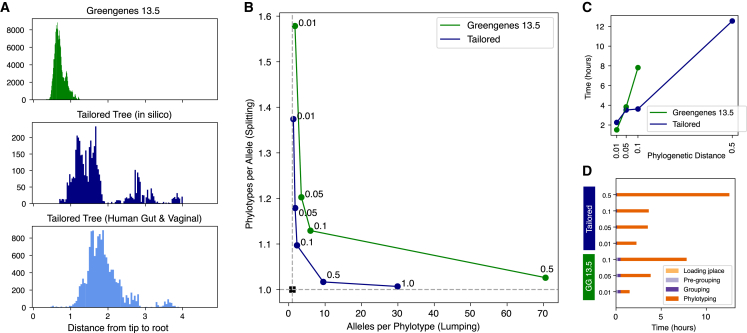
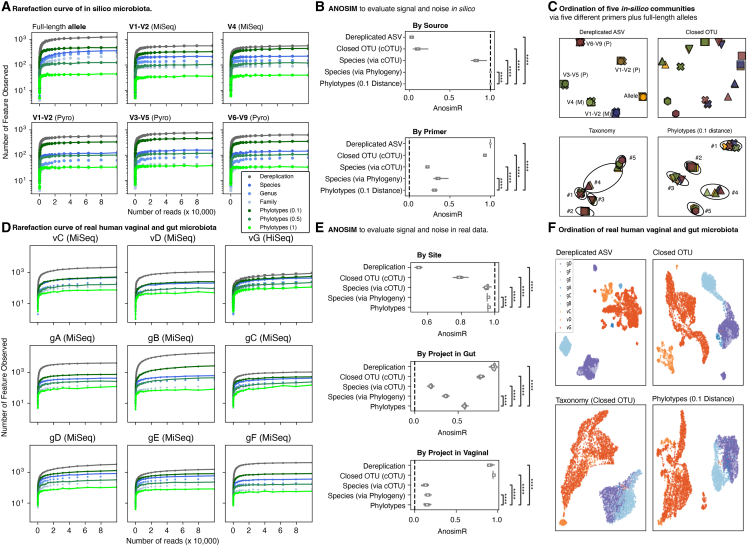
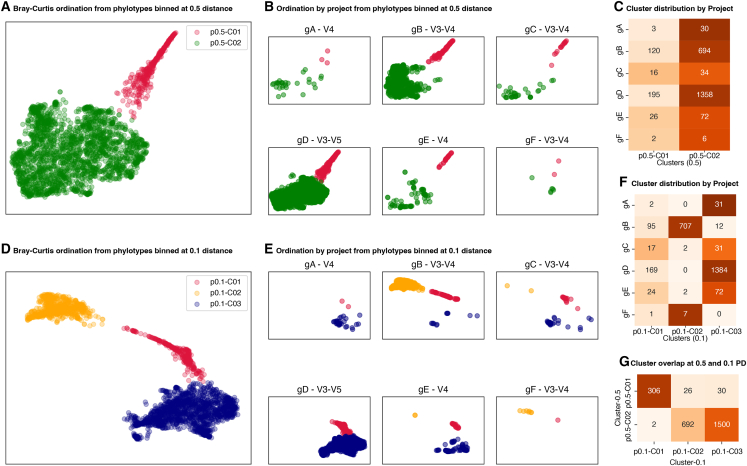
For studies using microbiome data, the ability to robustly combine data from technically and biologically distinct microbiome studies is a crucial means of supporting more robust and clinically relevant inferences. Formidable technical challenges arise when attempting to combine data from technically diverse 16S rRNA gene variable region amplicon sequencing (16S) studies. Closed operational taxonomic units and taxonomy are criticized as being heavily dependent upon reference sets and with limited precision relative to the underlying biology. Phylogenetic placement has been demonstrated to be a promising taxonomy-free manner of harmonizing microbiome data, but it has lacked a validated count-based feature suitable for use in machine learning and association studies. Here we introduce a phylogenetic-placement-based, taxonomy-independent, compositional feature of microbiota: phylotypes. Phylotypes were predictive of clinical outcomes such as obesity or pre-term birth on technically diverse independent validation sets harmonized post hoc. Thus, phylotypes enable the rigorous cross-validation of 16S-based clinical prognostic models and associative microbiome studies.
对于使用微生物组数据的研究来说,稳健地组合来自技术和生物学上不同的微生物组研究的数据是支持更稳健和更具临床相关性推断的关键手段。当试图组合来自技术多样化的 16S rRNA 基因可变区扩增子测序(16S)研究的数据时,会出现令人生畏的技术挑战。封闭的操作分类单元和分类法被批评为严重依赖参考集,并且相对于基础生物学而言精度有限。系统发育定位已被证明是一种有前途的无分类学方法,可以协调微生物组数据,但它缺乏经过验证的基于计数的特征,不适合用于机器学习和关联研究。在这里,我们介绍一种基于系统发育定位、独立于分类学的微生物组组成特征:菌群。在技术上多样化的独立验证集上进行事后协调后,菌群可以预测肥胖或早产等临床结果。因此,菌群可以严格地对基于 16S 的临床预后模型和关联微生物组研究进行交叉验证。