Department of Computer Science and Engineering, Yuan Ze University, Taoyuan, 32003, Taiwan.
Professional Master Program in Artificial Intelligence in Medicine, College of Medicine, Taipei Medical University, Taipei City, 106, Taiwan.
BMC Med Genomics. 2020 Oct 22;13(Suppl 10):155. doi: 10.1186/s12920-020-00779-w.
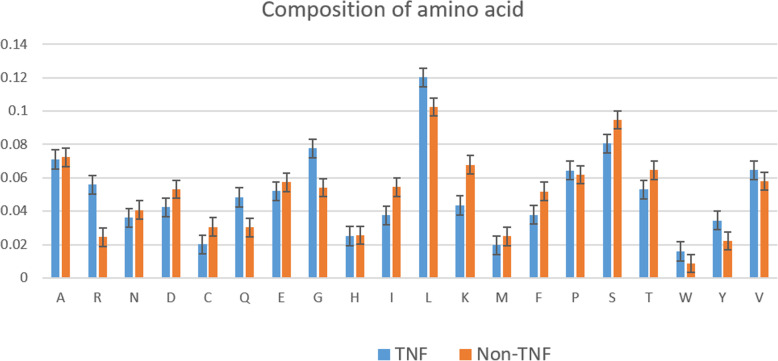
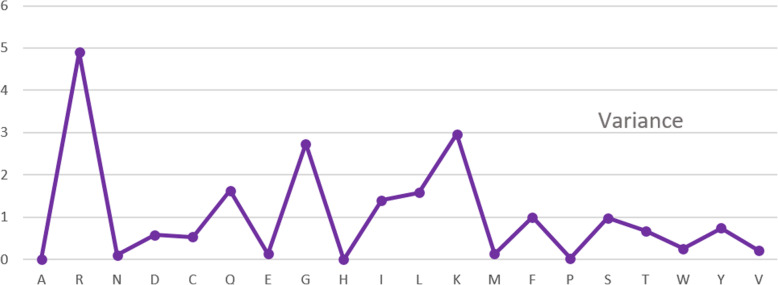
Cytokines are a class of small proteins that act as chemical messengers and play a significant role in essential cellular processes including immunity regulation, hematopoiesis, and inflammation. As one important family of cytokines, tumor necrosis factors have association with the regulation of a various biological processes such as proliferation and differentiation of cells, apoptosis, lipid metabolism, and coagulation. The implication of these cytokines can also be seen in various diseases such as insulin resistance, autoimmune diseases, and cancer. Considering the interdependence between this kind of cytokine and others, classifying tumor necrosis factors from other cytokines is a challenge for biological scientists.
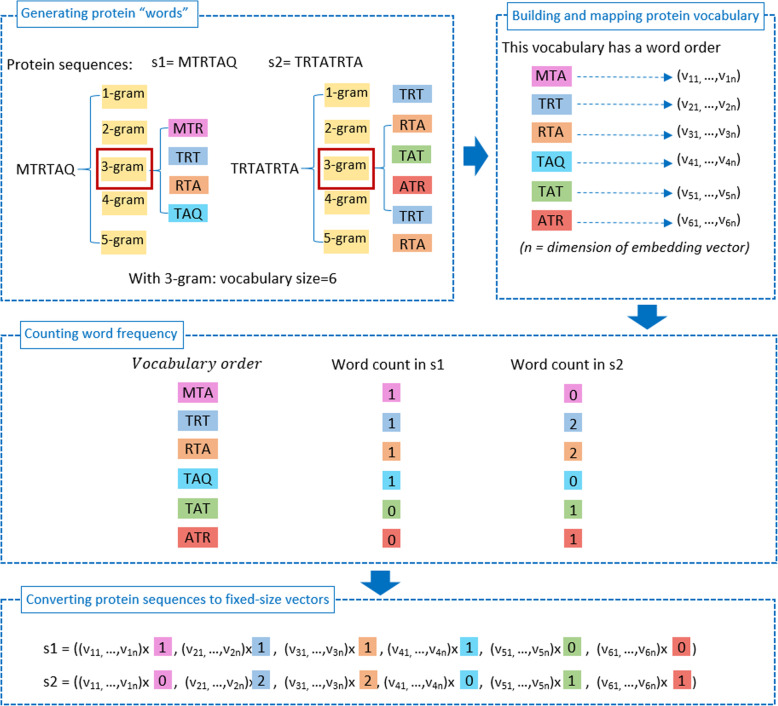
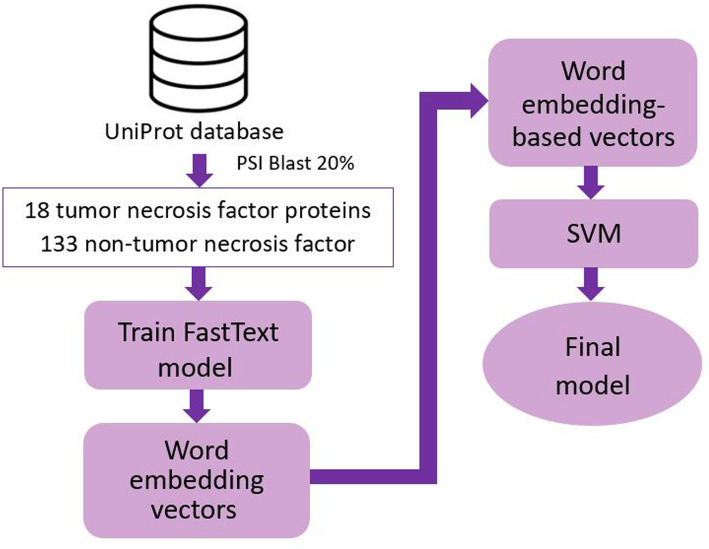
In this research, we employed a word embedding technique to create hybrid features which was proved to efficiently identify tumor necrosis factors given cytokine sequences. We segmented each protein sequence into protein words and created corresponding word embedding for each word. Then, word embedding-based vector for each sequence was created and input into machine learning classification models. When extracting feature sets, we not only diversified segmentation sizes of protein sequence but also conducted different combinations among split grams to find the best features which generated the optimal prediction. Furthermore, our methodology follows a well-defined procedure to build a reliable classification tool.
With our proposed hybrid features, prediction models obtain more promising performance compared to seven prominent sequenced-based feature kinds. Results from 10 independent runs on the surveyed dataset show that on an average, our optimal models obtain an area under the curve of 0.984 and 0.998 on 5-fold cross-validation and independent test, respectively.
These results show that biologists can use our model to identify tumor necrosis factors from other cytokines efficiently. Moreover, this study proves that natural language processing techniques can be applied reasonably to help biologists solve bioinformatics problems efficiently.
细胞因子是一类小分子蛋白质,作为化学信使,在包括免疫调节、造血和炎症在内的基本细胞过程中发挥重要作用。作为细胞因子的一个重要家族,肿瘤坏死因子与细胞的增殖和分化、细胞凋亡、脂代谢和凝血等各种生物过程的调节有关。这些细胞因子的意义也可以在胰岛素抵抗、自身免疫性疾病和癌症等各种疾病中看到。考虑到这种细胞因子与其他细胞因子之间的相互依存关系,将肿瘤坏死因子与其他细胞因子区分开来是生物科学家面临的一个挑战。
在这项研究中,我们采用了一种词嵌入技术来创建混合特征,事实证明,这种混合特征可以有效地识别细胞因子序列中的肿瘤坏死因子。我们将每个蛋白质序列分割成蛋白质单词,并为每个单词创建相应的词嵌入。然后,为每个序列创建基于词嵌入的向量,并将其输入到机器学习分类模型中。在提取特征集时,我们不仅多样化了蛋白质序列的分割大小,还在分割的单词之间进行了不同的组合,以找到产生最佳预测的最佳特征。此外,我们的方法遵循一个明确的步骤来构建一个可靠的分类工具。
使用我们提出的混合特征,与七种突出的基于序列的特征相比,预测模型的性能更有前景。在调查数据集上进行的 10 次独立运行的结果表明,平均而言,我们的最佳模型在 5 折交叉验证和独立测试中分别获得了 0.984 和 0.998 的曲线下面积。
这些结果表明,生物学家可以有效地使用我们的模型来识别其他细胞因子中的肿瘤坏死因子。此外,本研究证明了自然语言处理技术可以合理地应用于帮助生物学家有效地解决生物信息学问题。