Agricultural Biotechnology Centre, Faculty of Agriculture, University of Peradeniya, Peradeniya, 20400, Sri Lanka.
Postgraduate Institute of Science, University of Peradeniya, Peradeniya, 20400, Sri Lanka.
Sci Rep. 2020 Oct 26;10(1):18236. doi: 10.1038/s41598-020-75270-8.
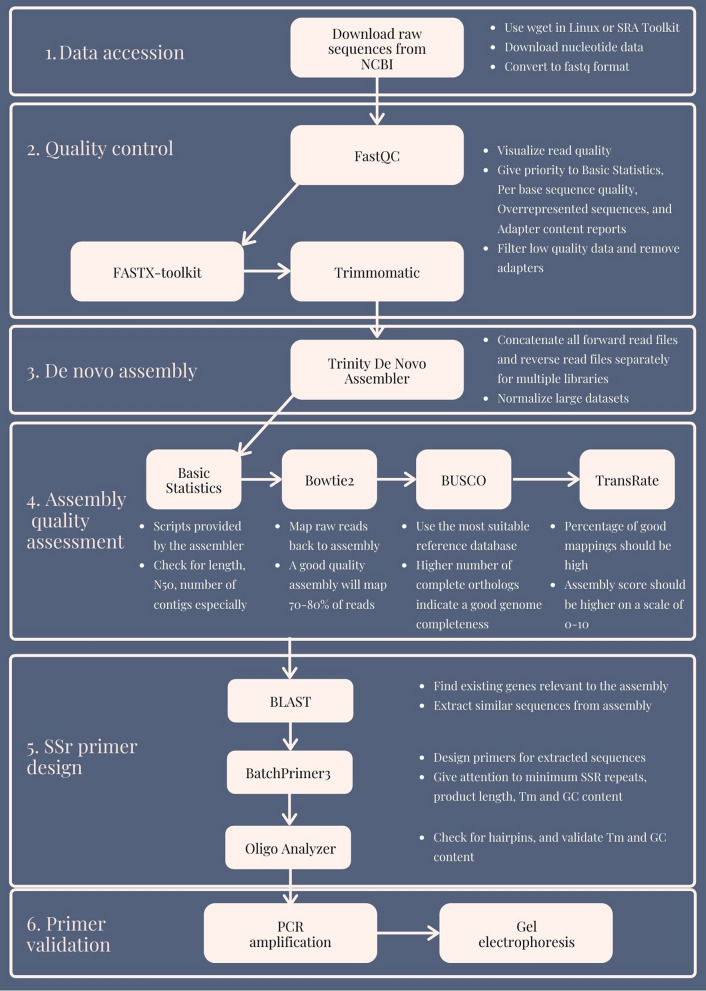
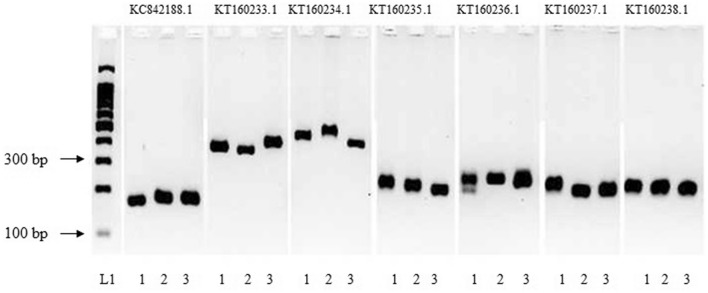
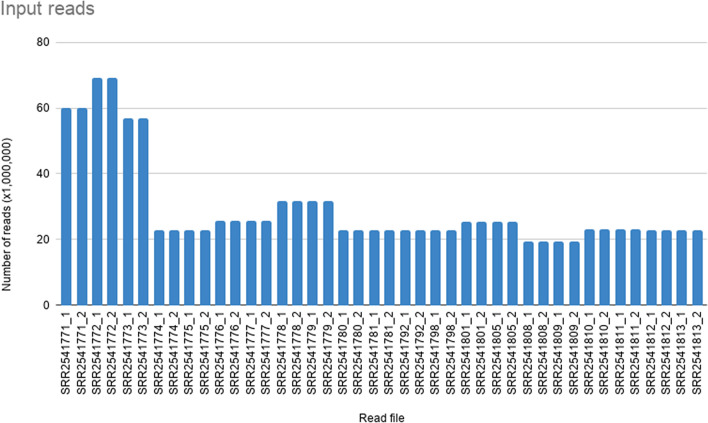
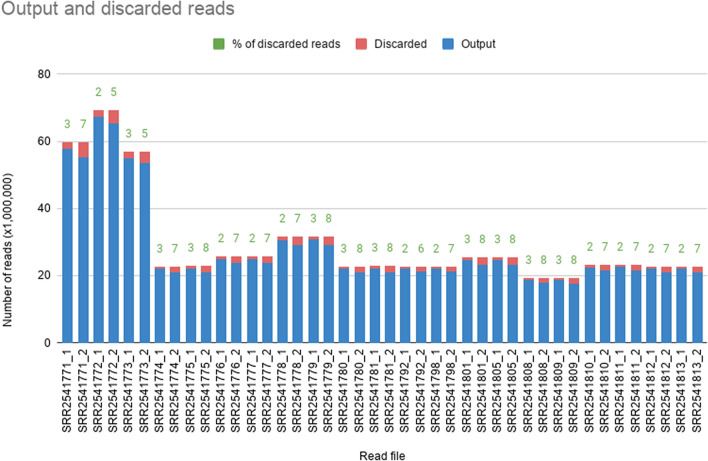
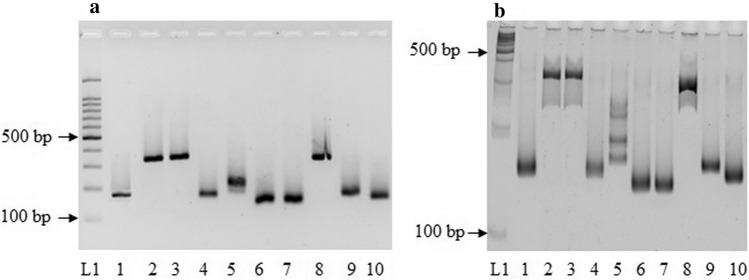
Recent advances in next-generation sequencing technologies have paved the path for a considerable amount of sequencing data at a relatively low cost. This has revolutionized the genomics and transcriptomics studies. However, different challenges are now created in handling such data with available bioinformatics platforms both in assembly and downstream analysis performed in order to infer correct biological meaning. Though there are a handful of commercial software and tools for some of the procedures, cost of such tools has made them prohibitive for most research laboratories. While individual open-source or free software tools are available for most of the bioinformatics applications, those components usually operate standalone and are not combined for a user-friendly workflow. Therefore, beginners in bioinformatics might find analysis procedures starting from raw sequence data too complicated and time-consuming with the associated learning-curve. Here, we outline a procedure for de novo transcriptome assembly and Simple Sequence Repeats (SSR) primer design solely based on tools that are available online for free use. For validation of the developed workflow, we used Illumina HiSeq reads of different tissue samples of Santalum album (sandalwood), generated from a previous transcriptomics project. A portion of the designed primers were tested in the lab with relevant samples and all of them successfully amplified the targeted regions. The presented bioinformatics workflow can accurately assemble quality transcriptomes and develop gene specific SSRs. Beginner biologists and researchers in bioinformatics can easily utilize this workflow for research purposes.
近年来,下一代测序技术的进步为相对较低的成本获得大量测序数据铺平了道路。这彻底改变了基因组学和转录组学研究。然而,现在在可用的生物信息学平台上处理这些数据时,无论是组装还是下游分析,都面临着不同的挑战,以便推断出正确的生物学意义。尽管有一些商业软件和工具可用于某些程序,但这些工具的成本使得它们对大多数研究实验室来说都是望尘莫及的。虽然大多数生物信息学应用都有一些开源或免费的软件工具,但这些组件通常是独立运行的,并没有为用户友好的工作流程进行组合。因此,生物信息学的初学者可能会发现,从原始序列数据开始的分析过程过于复杂和耗时,并且学习曲线也很陡峭。在这里,我们概述了一种仅基于免费在线工具的从头转录组组装和简单重复序列(SSR)引物设计的程序。为了验证开发的工作流程,我们使用了之前转录组学项目中不同组织样本的 Illumina HiSeq 读取数据。设计的一部分引物在实验室中用相关样本进行了测试,所有引物都成功地扩增了目标区域。所提出的生物信息学工作流程可以准确地组装高质量的转录组并开发基因特异性 SSR。生物信息学的初学者和研究人员可以轻松地将此工作流程用于研究目的。