Willems Patrick, Fijalkowski Igor, Van Damme Petra
Department of Biochemistry and Microbiology, Ghent University, Ghent, Belgium.
Department of Biochemistry and Microbiology, Ghent University, Ghent, Belgium
mSystems. 2020 Oct 27;5(5):e00833-20. doi: 10.1128/mSystems.00833-20.
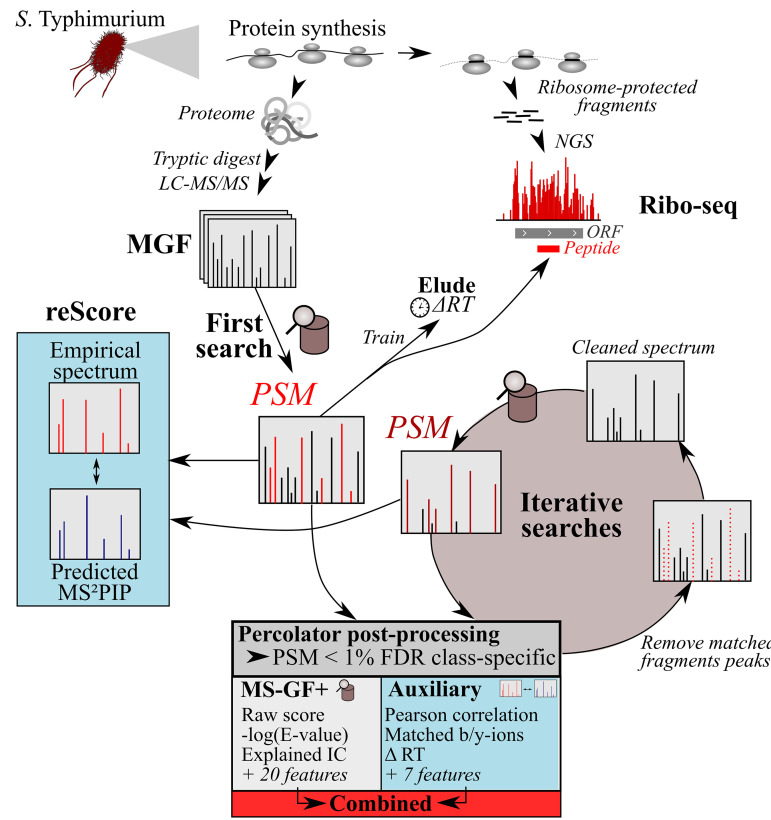
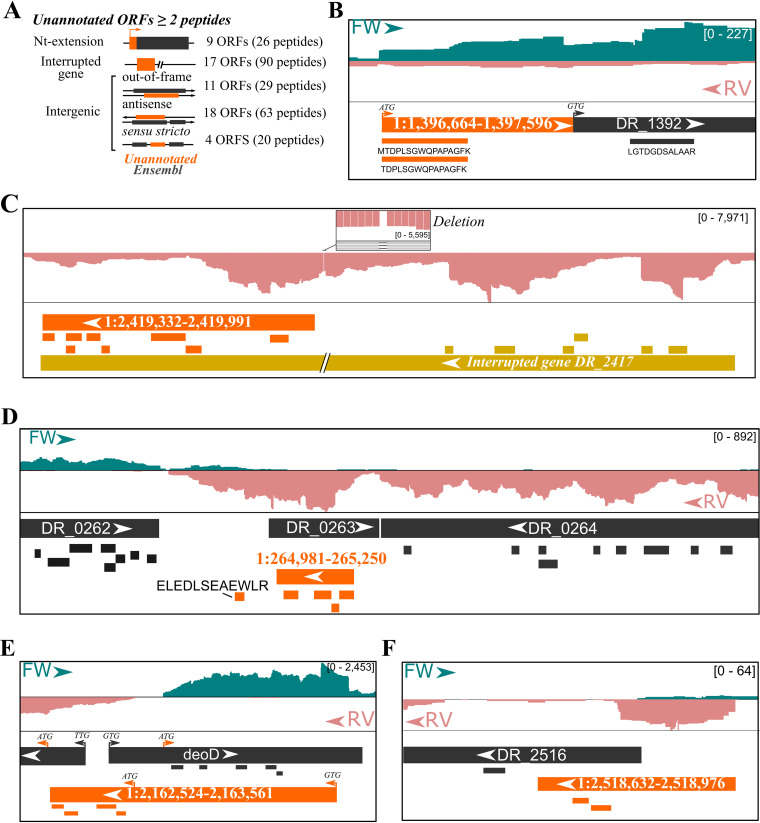
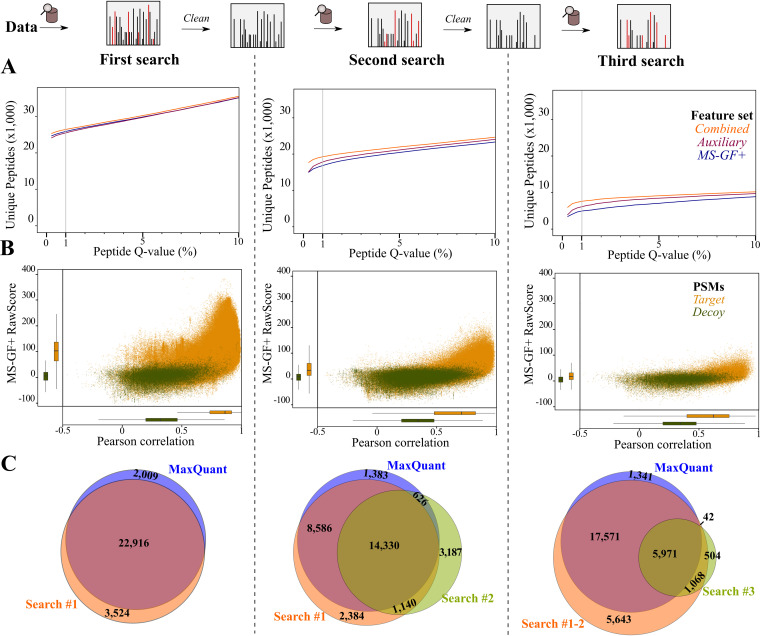
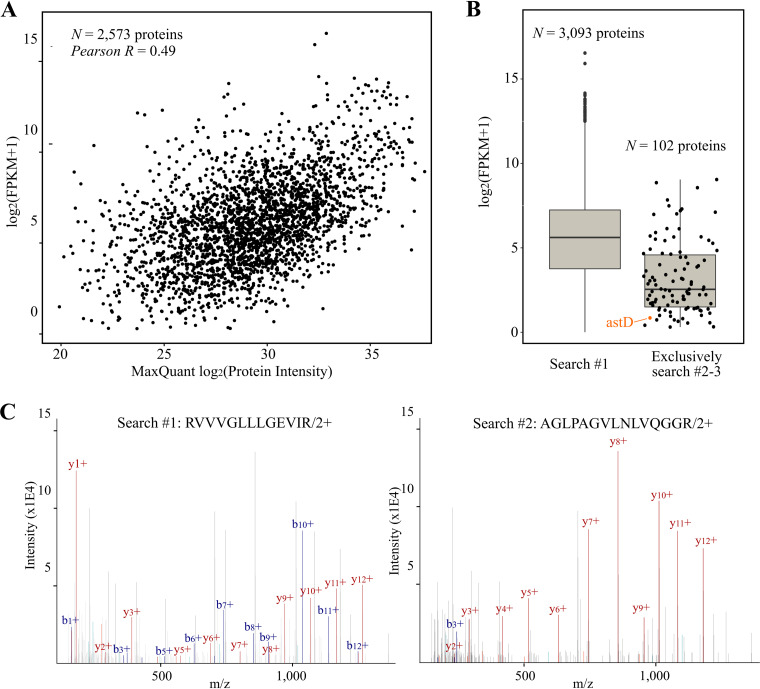
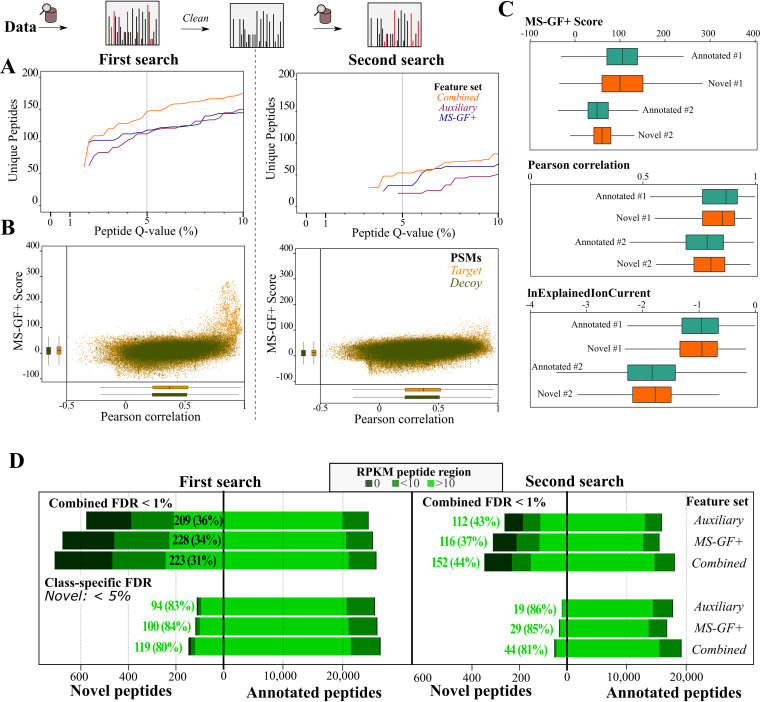
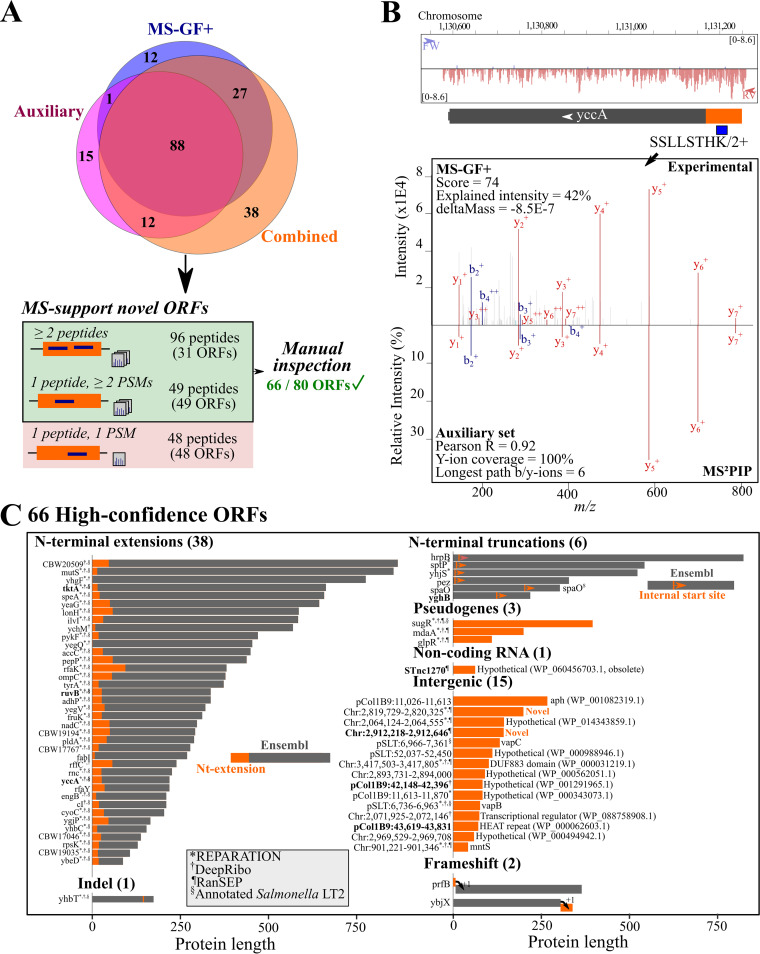
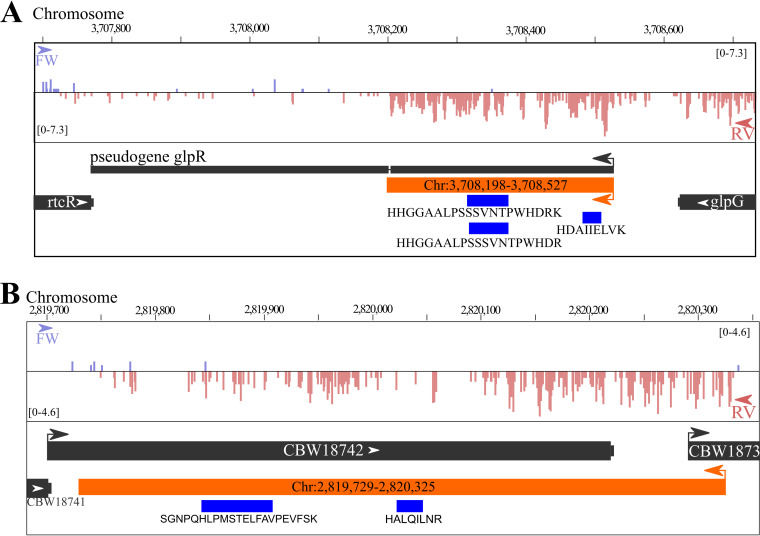
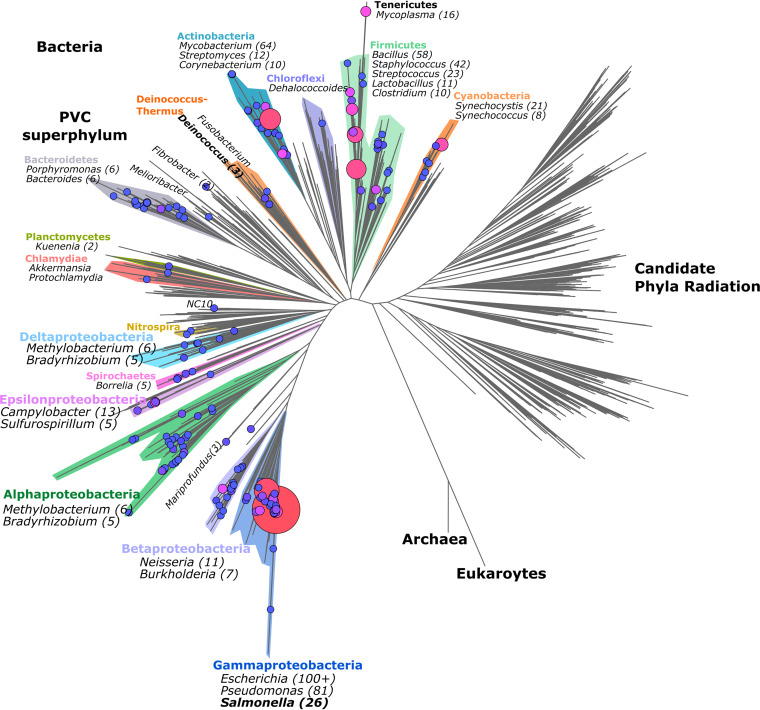
Prokaryotic genome annotation is heavily dependent on automated gene annotation pipelines that are prone to propagate errors and underestimate genome complexity. We describe an optimized proteogenomic workflow that uses ribosome profiling (ribo-seq) and proteomic data for serovar Typhimurium to identify unannotated proteins or alternative protein forms. This data analysis encompasses the searching of cofragmenting peptides and postprocessing with extended peptide-to-spectrum quality features, including comparison to predicted fragment ion intensities. When this strategy is applied, an enhanced proteome depth is achieved, as well as greater confidence for unannotated peptide hits. We demonstrate the general applicability of our pipeline by reanalyzing public data sets. Taken together, our results show that systematic reanalysis using available prokaryotic (proteome) data sets holds great promise to assist in experimentally based genome annotation. Delineation of open reading frames (ORFs) causes persistent inconsistencies in prokaryote genome annotation. We demonstrate that by advanced (re)analysis of omics data, a higher proteome coverage and sensitive detection of unannotated ORFs can be achieved, which can be exploited for conditional bacterial genome (re)annotation, which is especially relevant in view of annotating the wealth of sequenced prokaryotic genomes obtained in recent years.
原核生物基因组注释严重依赖于自动化基因注释流程,而这些流程容易传播错误并低估基因组复杂性。我们描述了一种优化的蛋白质基因组学工作流程,该流程使用鼠伤寒血清型的核糖体图谱分析(ribo-seq)和蛋白质组学数据来鉴定未注释的蛋白质或替代蛋白质形式。这种数据分析包括搜索共片段化肽段以及使用扩展的肽段与谱图质量特征进行后处理,包括与预测的碎片离子强度进行比较。当应用此策略时,可实现增强的蛋白质组深度,以及对未注释肽段匹配的更高置信度。我们通过重新分析公共数据集证明了我们流程的普遍适用性。综上所述,我们的结果表明,使用可用的原核生物(蛋白质组)数据集进行系统的重新分析有望极大地辅助基于实验的基因组注释。开放阅读框(ORF)的划定在原核生物基因组注释中导致了持续的不一致性。我们证明了通过对组学数据进行高级(重新)分析,可以实现更高的蛋白质组覆盖率和对未注释ORF的灵敏检测,并可将其用于条件性细菌基因组(重新)注释,鉴于近年来获得了大量已测序的原核生物基因组,这一点尤为重要。