Animal Breeding and Genomics, Wageningen University and Research, P.O. Box 338, 6700 AH, Wageningen, The Netherlands.
Centre for Genetic Resources the Netherlands, Wageningen University and Research, P.O. Box 338, 6700 AH, Wageningen, The Netherlands.
Genet Sel Evol. 2020 Oct 28;52(1):64. doi: 10.1186/s12711-020-00583-1.
Inbreeding depression refers to the decrease in mean performance due to inbreeding. Inbreeding depression is caused by an increase in homozygosity and reduced expression of (on average) favourable dominance effects. Dominance effects and allele frequencies differ across loci, and consequently inbreeding depression is expected to differ along the genome. In this study, we investigated differences in inbreeding depression across the genome of Dutch Holstein Friesian cattle, by estimating dominance effects and effects of regions of homozygosity (ROH).
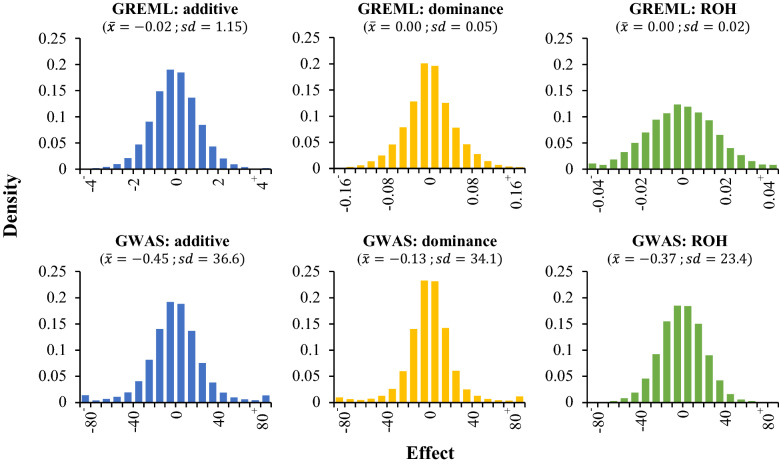
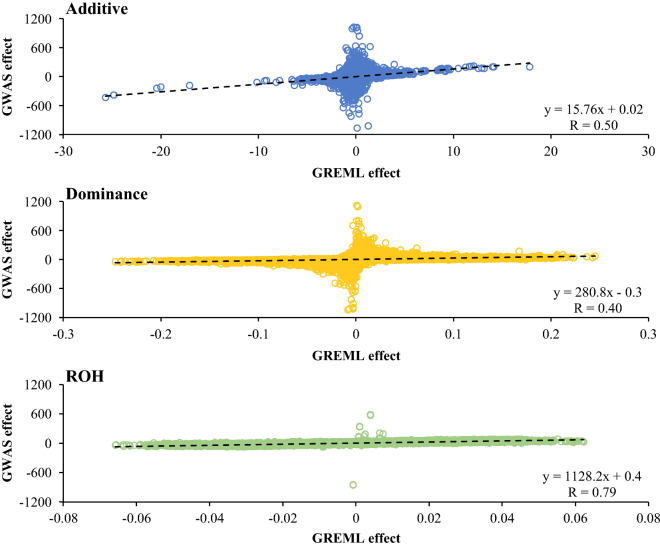
Genotype (75 k) and phenotype data of 38,792 cows were used. For nine yield, fertility and udder health traits, GREML models were run to estimate genome-wide inbreeding depression and estimate additive, dominance and ROH variance components. For this purpose, we introduced a ROH-based relationship matrix. Additive, dominance and ROH effects per SNP were obtained through back-solving. In addition, a single SNP GWAS was performed to identify significant additive, dominance or ROH associations.
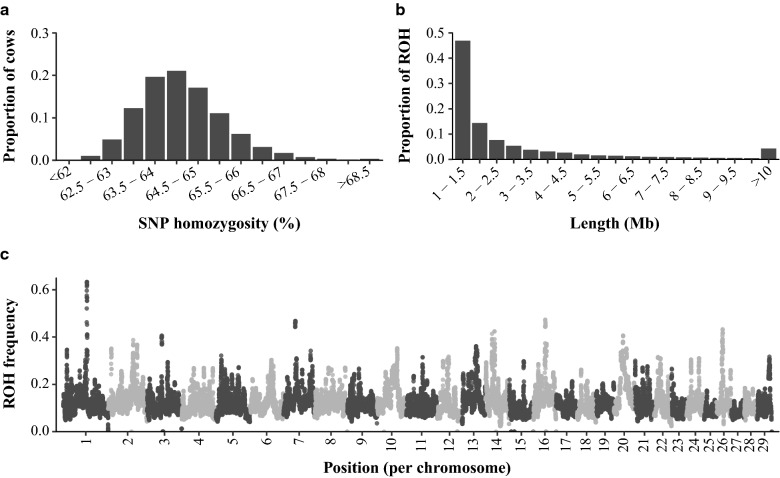
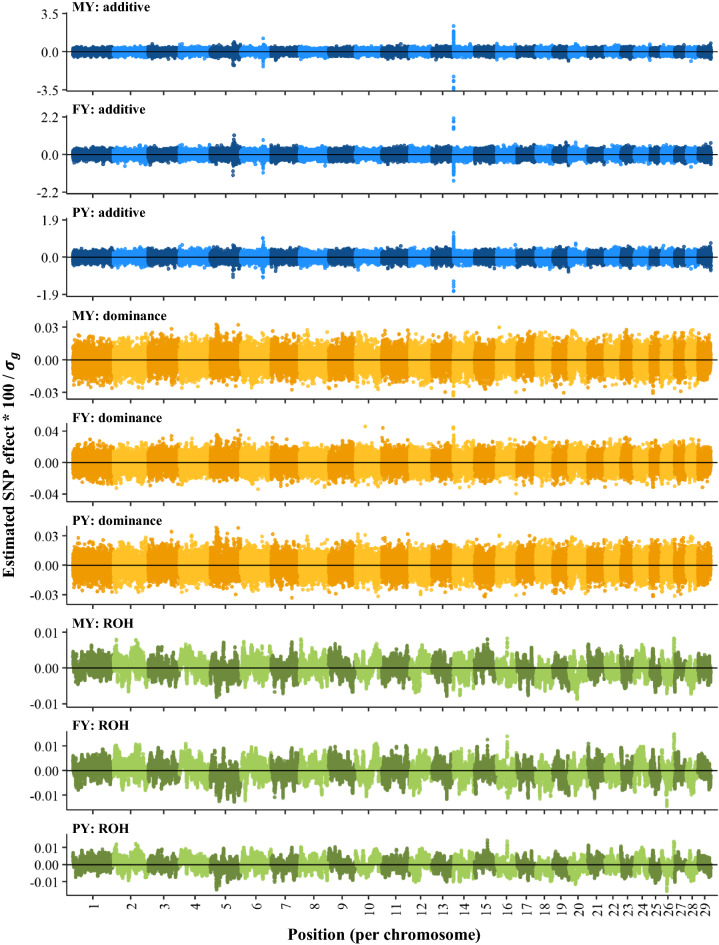
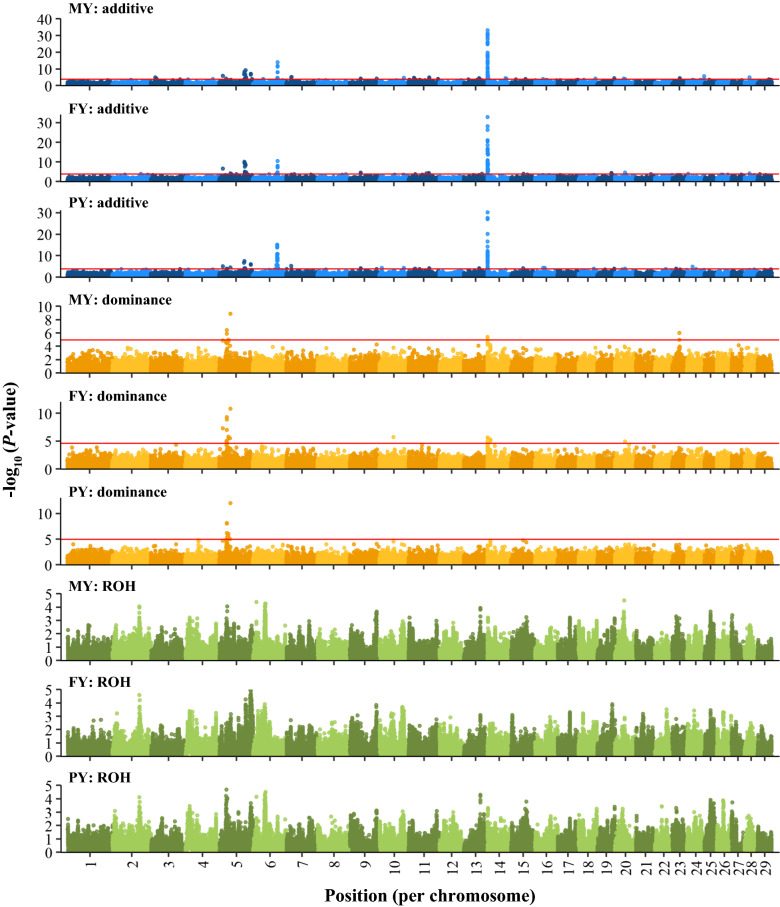
Genome-wide inbreeding depression was observed for all yield, fertility and udder health traits. For example, a 1% increase in genome-wide homozygosity was associated with a decrease in 305-d milk yield of approximately 99 kg. For yield traits only, including dominance and ROH effects in the GREML model resulted in a better fit (P < 0.05) than a model with only additive effects. After correcting for the effect of genome-wide homozygosity, dominance and ROH variance explained less than 1% of the phenotypic variance for all traits. Furthermore, dominance and ROH effects were distributed evenly along the genome. The most notable region with a favourable dominance effect for yield traits was on chromosome 5, but overall few regions with large favourable dominance effects and significant dominance associations were detected. No significant ROH-associations were found.
Inbreeding depression was distributed quite equally along the genome and was well captured by genome-wide homozygosity. These findings suggest that, based on 75 k SNP data, there is little benefit of accounting for region-specific inbreeding depression in selection schemes.
近交衰退是指由于近交导致的平均表现下降。近交衰退是由于杂合度增加和(平均而言)有利显性效应表达减少引起的。显性效应和等位基因频率在不同的基因座上有所不同,因此预期近交衰退会沿基因组而有所不同。在这项研究中,我们通过估计显性效应和纯合区域(ROH)的效应,研究了荷兰荷斯坦弗里生牛基因组中近交衰退的差异。
使用了 38792 头奶牛的基因型(75k)和表型数据。对于九个产奶量、繁殖力和乳房健康性状,我们运行 GREML 模型来估计全基因组近交衰退,并估计加性、显性和 ROH 方差分量。为此,我们引入了基于 ROH 的关系矩阵。通过反向求解获得每个 SNP 的加性、显性和 ROH 效应。此外,还进行了单 SNP GWAS,以鉴定显著的加性、显性或 ROH 关联。
所有产奶量、繁殖力和乳房健康性状都观察到全基因组近交衰退。例如,基因组范围内的杂合度增加 1%,与 305 天产奶量减少约 99kg 有关。仅对于产奶量性状,在 GREML 模型中包含显性和 ROH 效应会导致拟合更好(P<0.05),而仅包含加性效应的模型则不然。在校正全基因组范围内的杂合度效应后,显性和 ROH 方差解释的表型方差不到所有性状的 1%。此外,显性和 ROH 效应均匀分布在基因组上。对于产奶量性状,具有有利显性效应的最显著区域位于第 5 号染色体上,但总体上检测到的具有较大有利显性效应和显著显性关联的区域很少。没有发现显著的 ROH 关联。
近交衰退在基因组上分布相当均匀,并且很好地被全基因组杂合度所捕捉。这些发现表明,基于 75k SNP 数据,在选择方案中考虑特定区域的近交衰退几乎没有好处。