Klimm Florian, Toledo Enrique M, Monfeuga Thomas, Zhang Fang, Deane Charlotte M, Reinert Gesine
Department of Mathematics, Imperial College London, London, SW7 2AZ, UK.
Mitochondrial Biology Unit, University of Cambridge, Cambridge, CB2 0XY, UK.
BMC Genomics. 2020 Nov 2;21(1):756. doi: 10.1186/s12864-020-07144-2.
Recent advances in single-cell RNA sequencing have allowed researchers to explore transcriptional function at a cellular level. In particular, single-cell RNA sequencing reveals that there exist clusters of cells with similar gene expression profiles, representing different transcriptional states.
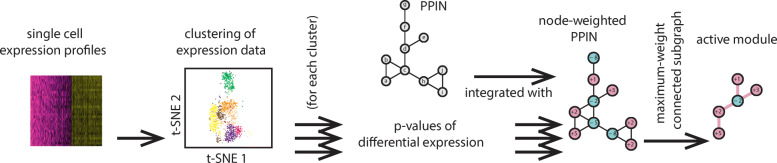
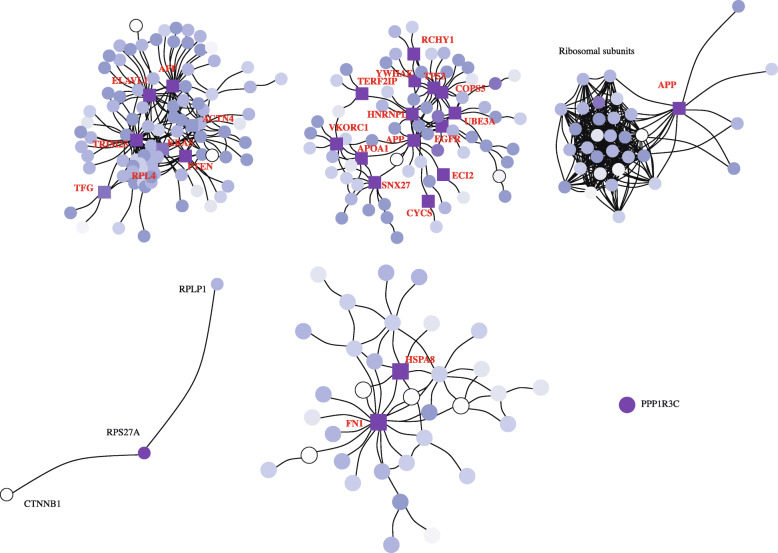
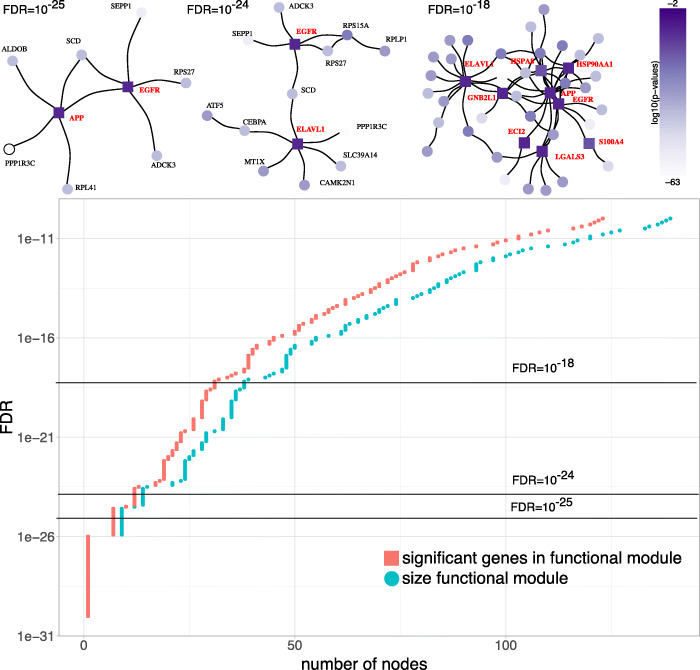
In this study, we present SCPPIN, a method for integrating single-cell RNA sequencing data with protein-protein interaction networks that detects active modules in cells of different transcriptional states. We achieve this by clustering RNA-sequencing data, identifying differentially expressed genes, constructing node-weighted protein-protein interaction networks, and finding the maximum-weight connected subgraphs with an exact Steiner-tree approach. As case studies, we investigate two RNA-sequencing data sets from human liver spheroids and human adipose tissue, respectively. With SCPPIN we expand the output of differential expressed genes analysis with information from protein interactions. We find that different transcriptional states have different subnetworks of the protein-protein interaction networks significantly enriched which represent biological pathways. In these pathways, SCPPIN identifies proteins that are not differentially expressed but have a crucial biological function (e.g., as receptors) and therefore reveals biology beyond a standard differential expressed gene analysis.
The introduced SCPPIN method can be used to systematically analyse differentially expressed genes in single-cell RNA sequencing data by integrating it with protein interaction data. The detected modules that characterise each cluster help to identify and hypothesise a biological function associated to those cells. Our analysis suggests the participation of unexpected proteins in these pathways that are undetectable from the single-cell RNA sequencing data alone. The techniques described here are applicable to other organisms and tissues.
单细胞RNA测序的最新进展使研究人员能够在细胞水平上探索转录功能。特别是,单细胞RNA测序揭示存在具有相似基因表达谱的细胞簇,代表不同的转录状态。
在本研究中,我们提出了SCPPIN,一种将单细胞RNA测序数据与蛋白质-蛋白质相互作用网络整合的方法,该方法可检测不同转录状态细胞中的活性模块。我们通过对RNA测序数据进行聚类、识别差异表达基因、构建节点加权蛋白质-蛋白质相互作用网络以及使用精确的斯坦纳树方法找到最大权重连通子图来实现这一目标。作为案例研究,我们分别研究了来自人肝球体和人脂肪组织的两个RNA测序数据集。使用SCPPIN,我们利用蛋白质相互作用的信息扩展了差异表达基因分析的输出。我们发现不同的转录状态具有蛋白质-蛋白质相互作用网络中显著富集的不同子网,这些子网代表生物学途径。在这些途径中,SCPPIN识别出未差异表达但具有关键生物学功能(例如作为受体)的蛋白质,因此揭示了超出标准差异表达基因分析的生物学信息。
引入的SCPPIN方法可用于通过将单细胞RNA测序数据与蛋白质相互作用数据整合来系统地分析其中的差异表达基因。表征每个簇的检测到的模块有助于识别和推测与这些细胞相关的生物学功能。我们的分析表明意外蛋白质参与了这些仅从单细胞RNA测序数据中无法检测到的途径。这里描述的技术适用于其他生物体和组织。