Institute of Bioinformatics, University of Georgia, 120 Green St., Athens, GA, 30602, USA.
Department of Statistics, University of Georgia, 310 Herty Drive, Athens, GA, 30602, USA.
BMC Bioinformatics. 2020 Nov 12;21(1):520. doi: 10.1186/s12859-020-03842-6.
Protein kinases are a large family of druggable proteins that are genomically and proteomically altered in many human cancers. Kinase-targeted drugs are emerging as promising avenues for personalized medicine because of the differential response shown by altered kinases to drug treatment in patients and cell-based assays. However, an incomplete understanding of the relationships connecting genome, proteome and drug sensitivity profiles present a major bottleneck in targeting kinases for personalized medicine.
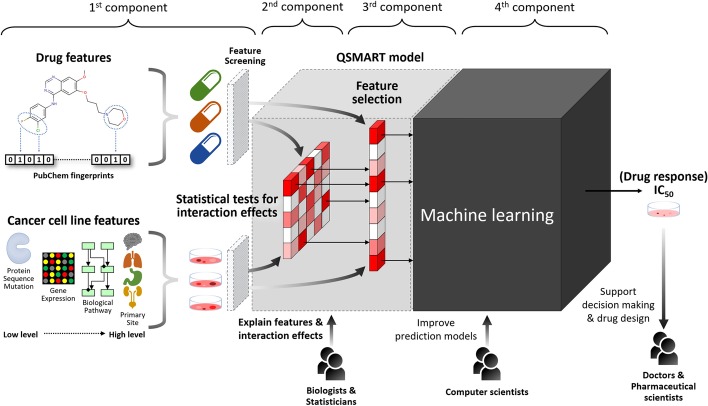
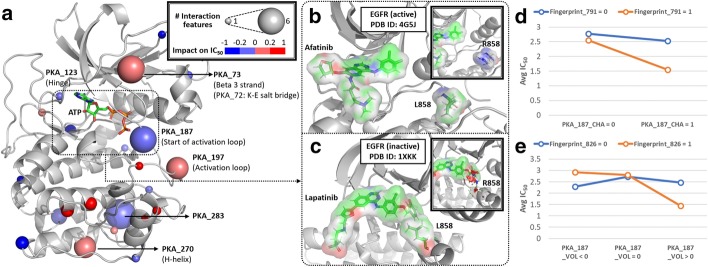
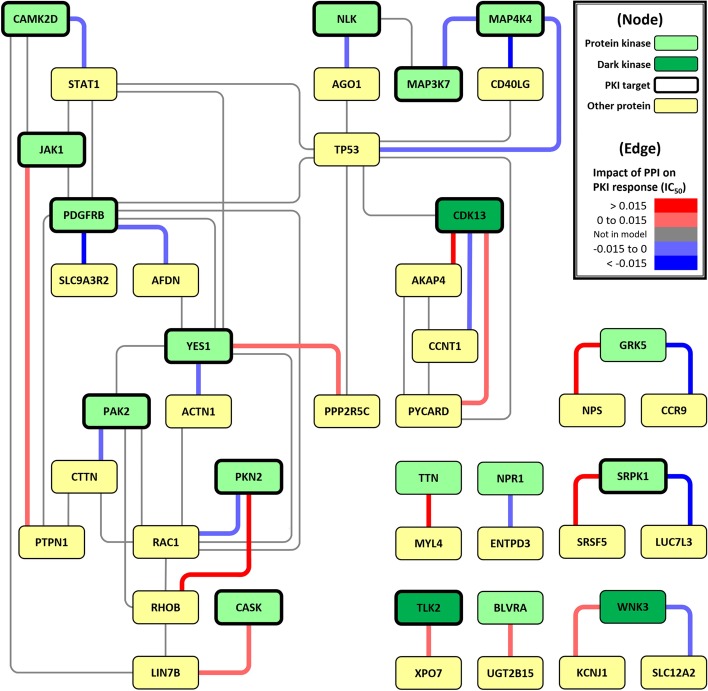
In this study, we propose a multi-component Quantitative Structure-Mutation-Activity Relationship Tests (QSMART) model and neural networks framework for providing explainable models of protein kinase inhibition and drug response ([Formula: see text]) profiles in cell lines. Using non-small cell lung cancer as a case study, we show that interaction terms that capture associations between drugs, pathways, and mutant kinases quantitatively contribute to the response of two EGFR inhibitors (afatinib and lapatinib). In particular, protein-protein interactions associated with the JNK apoptotic pathway, associations between lung development and axon extension, and interaction terms connecting drug substructures and the volume/charge of mutant residues at specific structural locations contribute significantly to the observed [Formula: see text] values in cell-based assays.
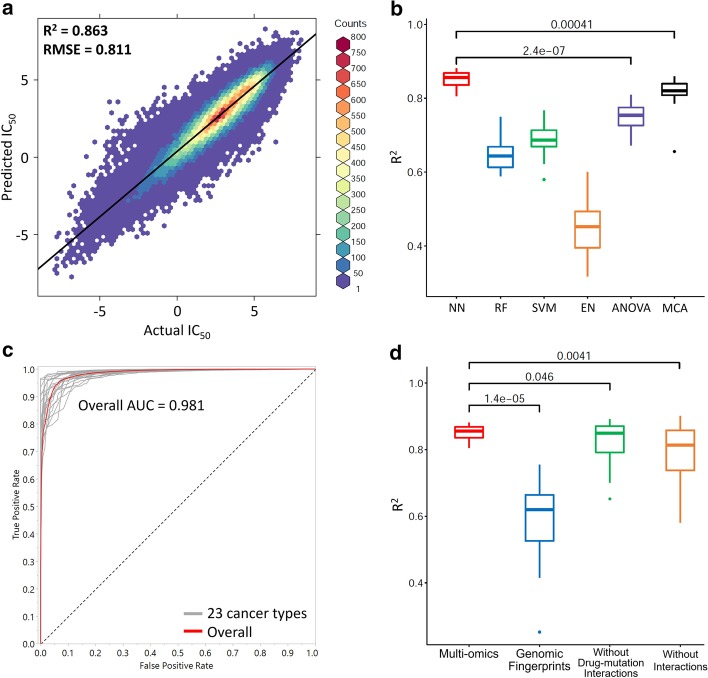
By integrating multi-omics data in the QSMART model, we not only predict drug responses in cancer cell lines with high accuracy but also identify features and explainable interaction terms contributing to the accuracy. Although we have tested our multi-component explainable framework on protein kinase inhibitors, it can be extended across the proteome to investigate the complex relationships connecting genotypes and drug sensitivity profiles.
蛋白激酶是一个庞大的可成药蛋白家族,在许多人类癌症中发生了基因组和蛋白质组改变。由于改变的激酶对患者和基于细胞的测定中药物治疗的反应不同,激酶靶向药物作为个性化医学的有前途途径正在出现。然而,对连接基因组、蛋白质组和药物敏感性谱的关系的不完全理解是针对个性化医学靶向激酶的主要瓶颈。
在这项研究中,我们提出了一种多成分定量结构-突变-活性关系测试(QSMART)模型和神经网络框架,用于提供蛋白激酶抑制和药物反应[Formula: see text]在细胞系中的可解释模型。使用非小细胞肺癌作为案例研究,我们表明,捕获药物、途径和突变激酶之间关联的相互作用项定量地有助于两种 EGFR 抑制剂(阿法替尼和拉帕替尼)的反应。特别是,与 JNK 凋亡途径相关的蛋白质-蛋白质相互作用、肺发育和轴突延伸之间的关联,以及连接药物亚结构和特定结构位置突变残基的体积/电荷的相互作用项,对细胞基础测定中观察到的[Formula: see text]值有显著贡献。
通过在 QSMART 模型中整合多组学数据,我们不仅可以高精度地预测癌症细胞系中的药物反应,而且可以确定有助于准确性的特征和可解释的相互作用项。虽然我们已经在蛋白激酶抑制剂上测试了我们的多成分可解释框架,但它可以扩展到整个蛋白质组,以研究连接基因型和药物敏感性谱的复杂关系。