School of Computer Science and Engineering, Central South University, Changsha, 410075, China.
School of Computer and Data Science, Henan University of Urban Construction, Pingdingshan, 467000, China.
BMC Bioinformatics. 2020 Nov 12;21(1):519. doi: 10.1186/s12859-020-03748-3.
Circular RNAs (circRNAs) are special noncoding RNA molecules with closed loop structures. Compared with the traditional linear RNA, circRNA is more stable and not easily degraded. Many studies have shown that circRNAs are involved in the regulation of various diseases and cancers. Determining the functions of circRNAs in mammalian cells is of great significance for revealing their mechanism of action in physiological and pathological processes, diagnosis and treatment of diseases. However, determining the functions of circRNAs on a large scale is a challenging task because of the high experimental costs.
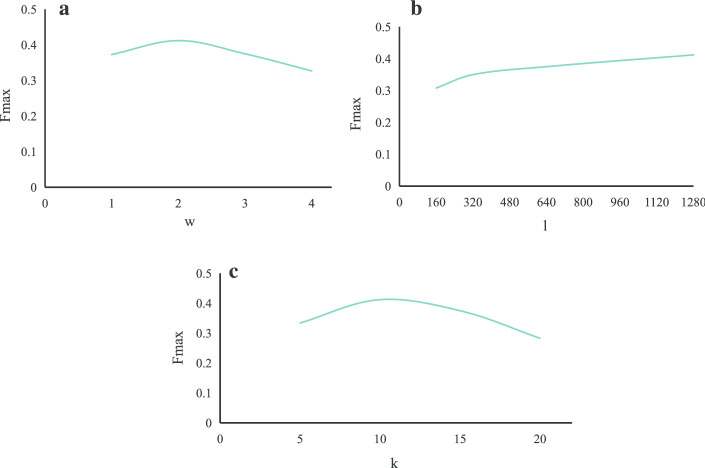
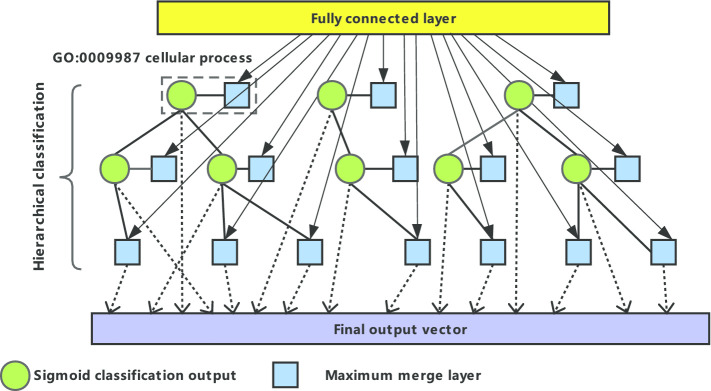
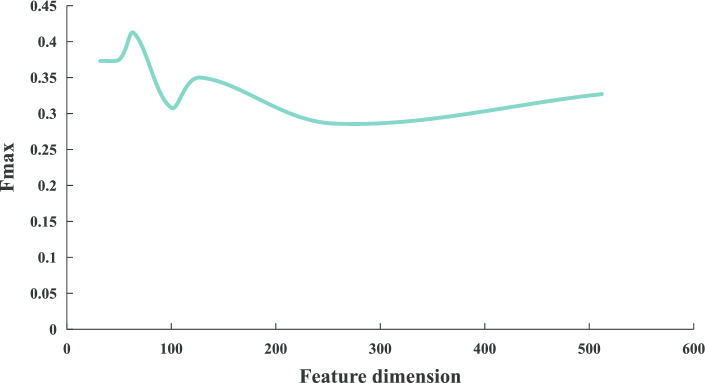
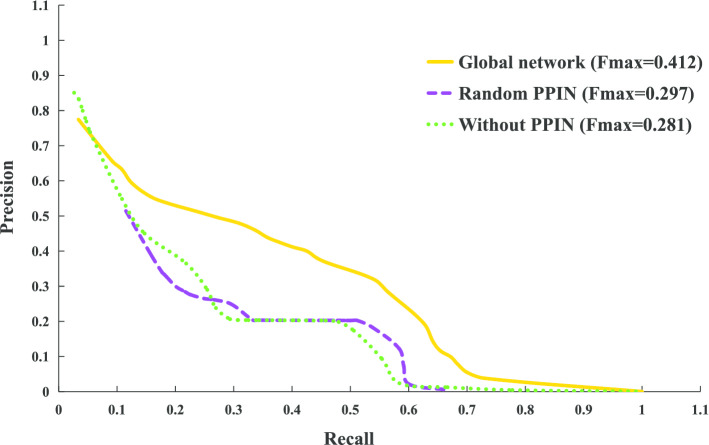
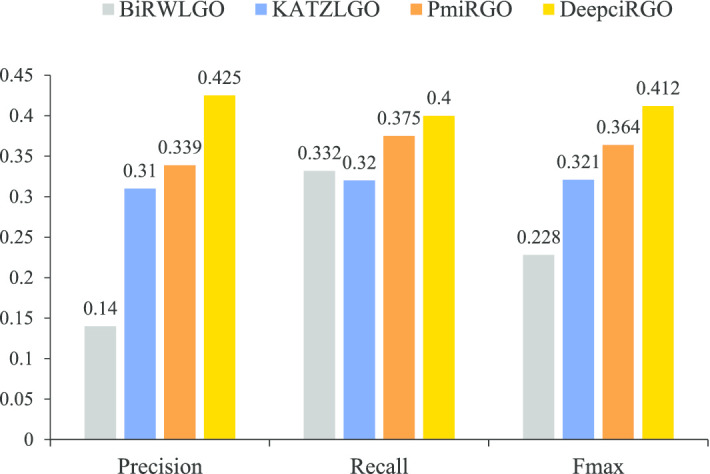
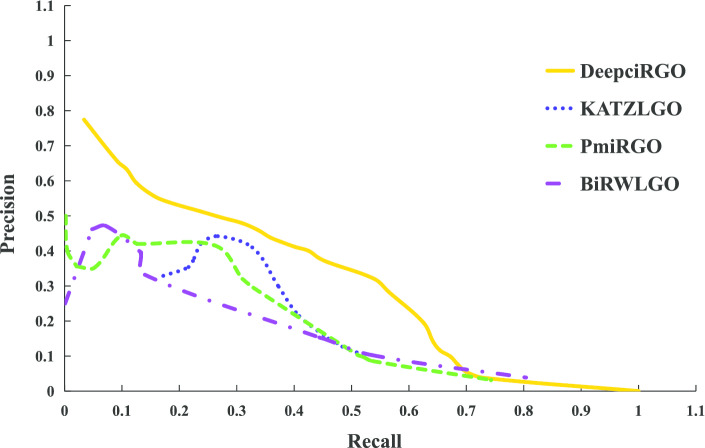
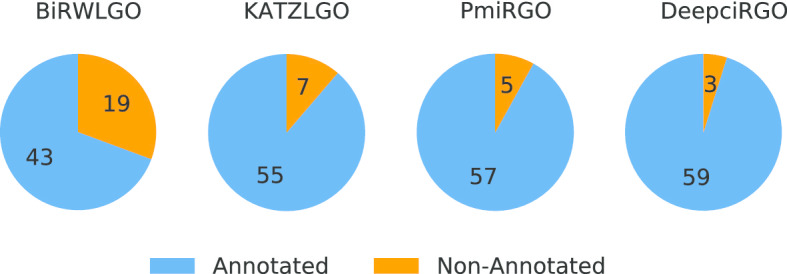
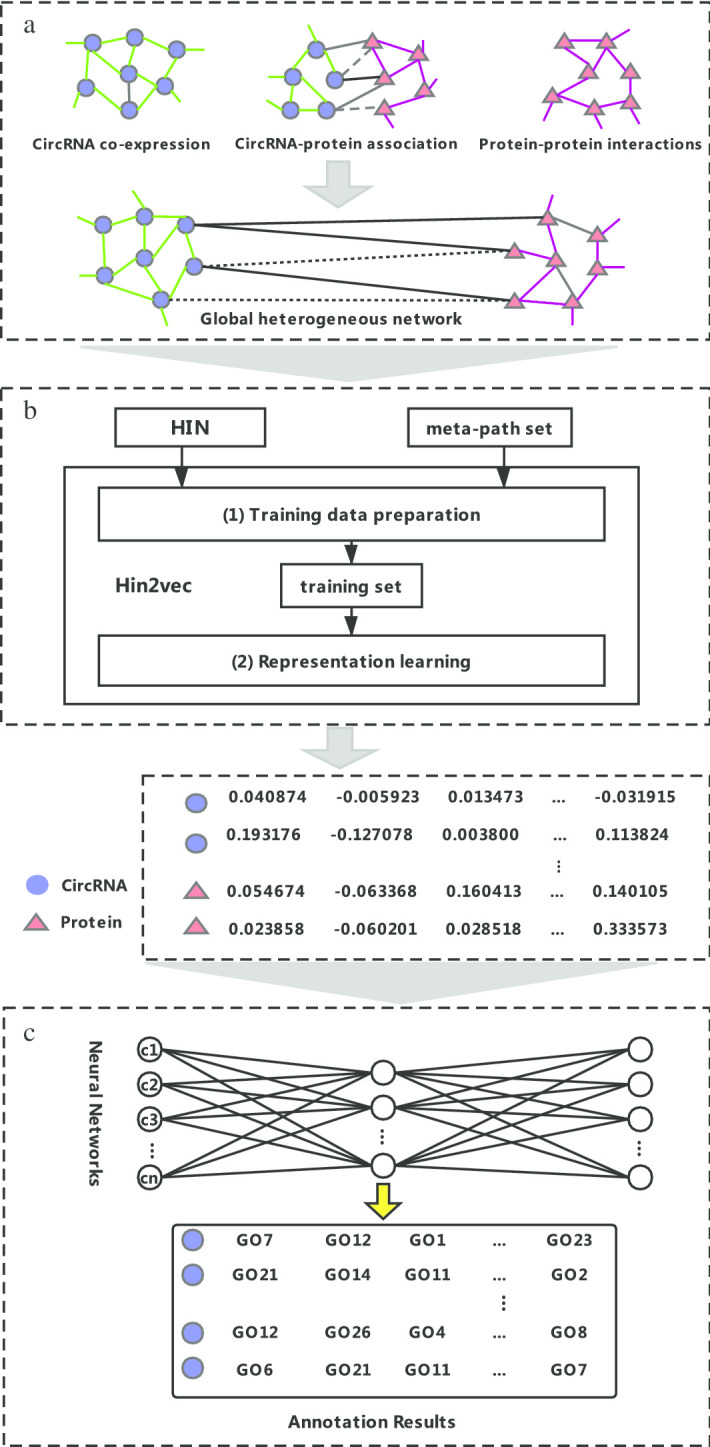
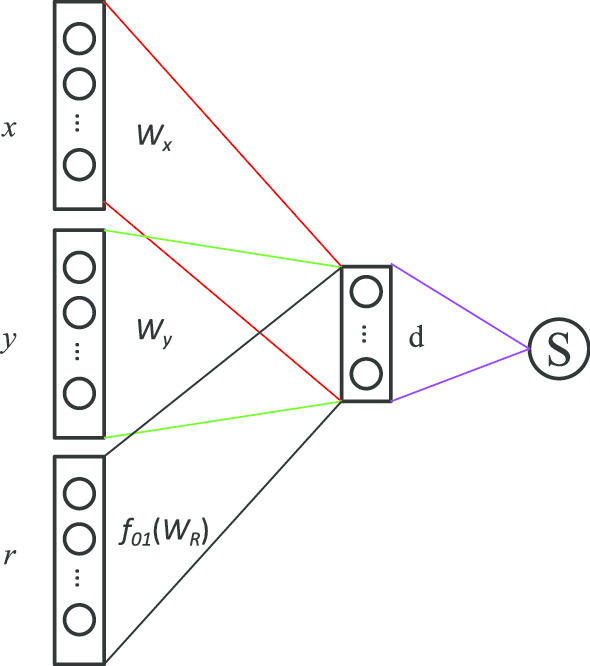
In this paper, we present a hierarchical deep learning model, DeepciRGO, which can effectively predict gene ontology functions of circRNAs. We build a heterogeneous network containing circRNA co-expressions, protein-protein interactions and protein-circRNA interactions. The topology features of proteins and circRNAs are calculated using a novel representation learning approach HIN2Vec across the heterogeneous network. Then, a deep multi-label hierarchical classification model is trained with the topology features to predict the biological process function in the gene ontology for each circRNA. In particular, we manually curated a benchmark dataset containing 185 GO annotations for 62 circRNAs, namely, circRNA2GO-62. The DeepciRGO achieves promising performance on the circRNA2GO-62 dataset with a maximum F-measure of 0.412, a recall score of 0.400, and an accuracy of 0.425, which are significantly better than other state-of-the-art RNA function prediction methods. In addition, we demonstrate the considerable potential of integrating multiple interactions and association networks.
DeepciRGO will be a useful tool for accurately annotating circRNAs. The experimental results show that integrating multi-source data can help to improve the predictive performance of DeepciRGO. Moreover, The model also can combine RNA structure and sequence information to further optimize predictive performance.
Circular RNAs (circRNAs) 是具有封闭环结构的特殊非编码 RNA 分子。与传统的线性 RNA 相比,circRNA 更稳定,不易降解。许多研究表明,circRNAs 参与了各种疾病和癌症的调控。确定 circRNAs 在哺乳动物细胞中的功能对于揭示它们在生理和病理过程中的作用机制、疾病的诊断和治疗具有重要意义。然而,由于实验成本高,大规模确定 circRNAs 的功能是一项具有挑战性的任务。
在本文中,我们提出了一种分层深度学习模型 DeepciRGO,它可以有效地预测 circRNAs 的基因本体功能。我们构建了一个包含 circRNA 共表达、蛋白质-蛋白质相互作用和蛋白质-circRNA 相互作用的异质网络。使用一种新的表示学习方法 HIN2Vec 在异质网络中计算蛋白质和 circRNA 的拓扑特征。然后,使用拓扑特征训练深度多标签层次分类模型,以预测每个 circRNA 在基因本体中的生物过程功能。特别是,我们手动整理了一个包含 62 个 circRNA 的 185 个 GO 注释的基准数据集,即 circRNA2GO-62。DeepciRGO 在 circRNA2GO-62 数据集上的表现非常出色,最大 F1 值为 0.412,召回率为 0.400,准确率为 0.425,明显优于其他最先进的 RNA 功能预测方法。此外,我们还证明了整合多种相互作用和关联网络的相当大的潜力。
DeepciRGO 将成为准确注释 circRNAs 的有用工具。实验结果表明,整合多源数据可以帮助提高 DeepciRGO 的预测性能。此外,该模型还可以结合 RNA 结构和序列信息,进一步优化预测性能。