Bioinformatics and Molecular Biology Unit, Department of Biochemistry, Federal University of Technology Akure, Ondo, Nigeria.
Phytomedicine Biochemical Pharmacology and Toxicology Unit, Department of Biochemistry, Federal University of Technology Akure, Ondo, Nigeria.
Mol Divers. 2021 Aug;25(3):1761-1773. doi: 10.1007/s11030-020-10151-w. Epub 2020 Nov 17.
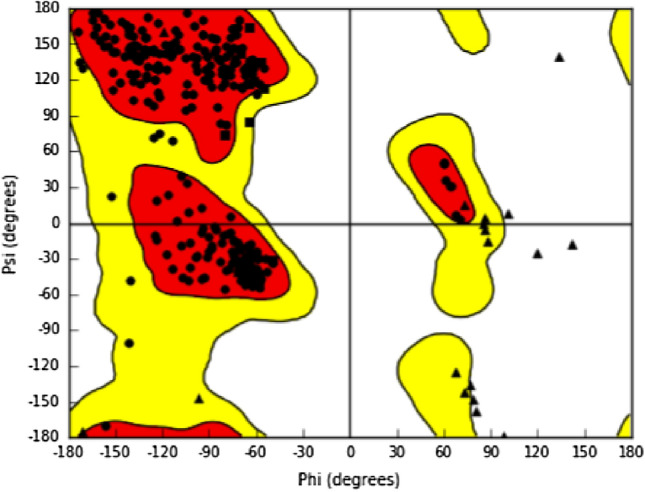
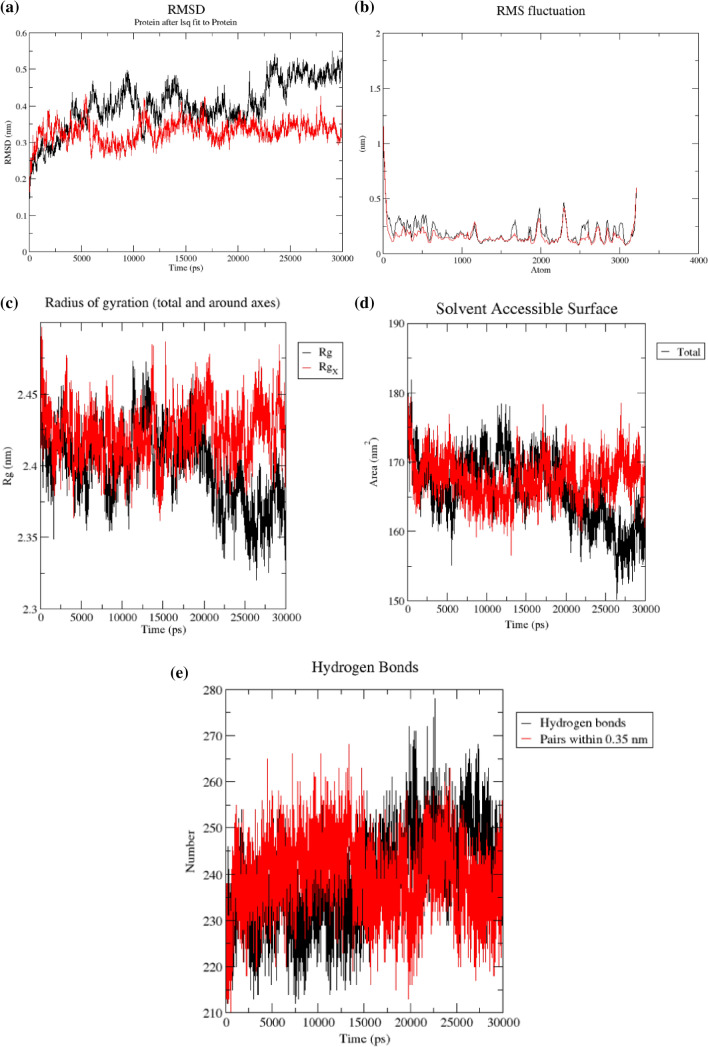
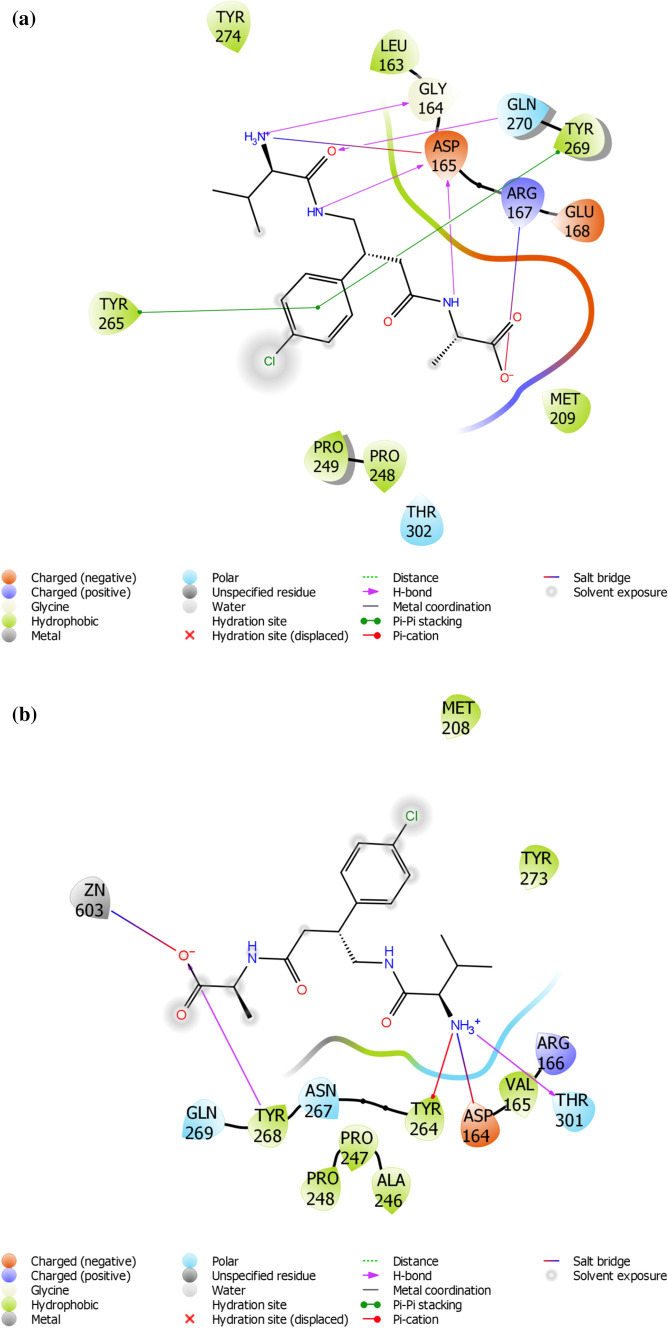
SARS-CoV-2 is a new strain of Coronavirus that caused the pneumonia outbreak in Wuhan, China and has spread to over 200 countries of the world. It has received worldwide attention due to its virulence and high rate of infection. So far, several drugs have experimented against SARS-CoV-2, but the failure of these drugs to specifically interact with the viral protease necessitates urgent measure to boost up researches for the development of effective therapeutics against SARS-CoV-2. Papain-like protease (PLpro) of the viral polyproteins is essential for maturation and infectivity of the virus, making it one of the prime targets explored for SARS-CoV-2 drug design. This study was conducted to evaluate the efficacy of ~ 50,000 natural compounds retrieved from IBS database against COVID-19 PLpro using computer-aided drug design. Based on molecular dock scores, molecular interaction with active catalytic residues and molecular dynamics (MD) simulations studies, STOCK1N-69160 [(S)-2-((R)-4-((R)-2-amino-3-methylbutanamido)-3-(4-chlorophenyl) butanamido) propanoic acid hydrochloride] has been proposed as a novel inhibitor against COVID-19 PLpro. It demonstrated favourable docking score, the free energy of binding, interacted with key amino acid residues necessary for PLpro inhibition and also showed significant moderation for parameters investigated for ADME/tox (Adsorption, distribution, metabolism, excretion and toxicological) properties. The edge of the compound was further established by its stability in MD simulation conducted for 30 ns employing GROMACS software. We propose that STOCK1N-69160 is worth further investigation for preventing SARS-CoV-2.
SARS-CoV-2 是一种新型冠状病毒,导致了中国武汉的肺炎疫情爆发,并已传播到世界 200 多个国家。由于其毒性和高感染率,它引起了全球的关注。到目前为止,已经有几种药物针对 SARS-CoV-2 进行了实验,但这些药物未能与病毒蛋白酶特异性相互作用,因此迫切需要采取措施,加强对开发针对 SARS-CoV-2 的有效治疗方法的研究。病毒多蛋白中的木瓜蛋白酶样蛋白酶 (PLpro) 对病毒的成熟和感染性至关重要,使其成为探索 SARS-CoV-2 药物设计的主要靶点之一。本研究旨在利用计算机辅助药物设计,评估从 IBS 数据库中检索到的约 50,000 种天然化合物对 COVID-19 PLpro 的疗效。基于分子对接评分、与活性催化残基的分子相互作用以及分子动力学 (MD) 模拟研究,提出 STOCK1N-69160 [(S)-2-((R)-4-((R)-2-氨基-3-甲基丁酰胺基)-3-(4-氯苯基)丁酰胺基)丙酸盐酸盐] 作为一种新型 COVID-19 PLpro 抑制剂。它表现出良好的对接评分、结合自由能、与 PLpro 抑制所需的关键氨基酸残基相互作用,并且对 ADME/tox(吸收、分布、代谢、排泄和毒理学)特性的研究参数也表现出显著的调节作用。该化合物的边缘通过在 GROMACS 软件中进行 30 ns 的 MD 模拟进一步得到了证实。我们建议 STOCK1N-69160 值得进一步研究,以预防 SARS-CoV-2。