Computer Technologies Laboratory, ITMO University, Saint Petersburg, Russia.
JetBrains Research, Saint Petersburg, Russia.
BMC Bioinformatics. 2020 Nov 18;21(Suppl 6):261. doi: 10.1186/s12859-020-03572-9.
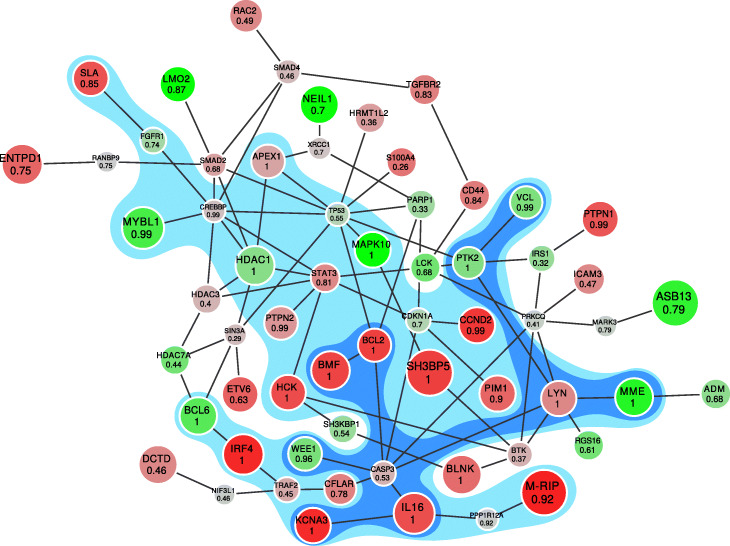
Integrative network methods are commonly used for interpretation of high-throughput experimental biological data: transcriptomics, proteomics, metabolomics and others. One of the common approaches is finding a connected subnetwork of a global interaction network that best encompasses significant individual changes in the data and represents a so-called active module. Usually methods implementing this approach find a single subnetwork and thus solve a hard classification problem for vertices. This subnetwork inherently contains erroneous vertices, while no instrument is provided to estimate the confidence level of any particular vertex inclusion. To address this issue, in the current study we consider the active module problem as a soft classification problem.
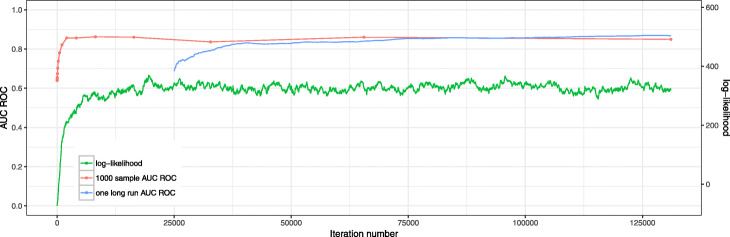
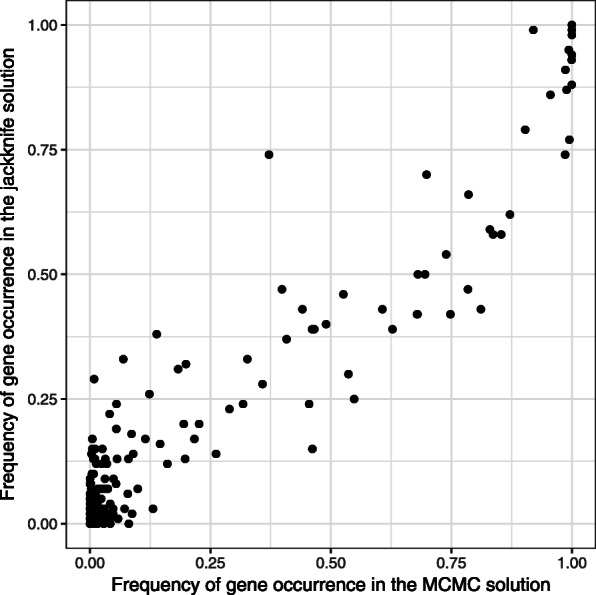
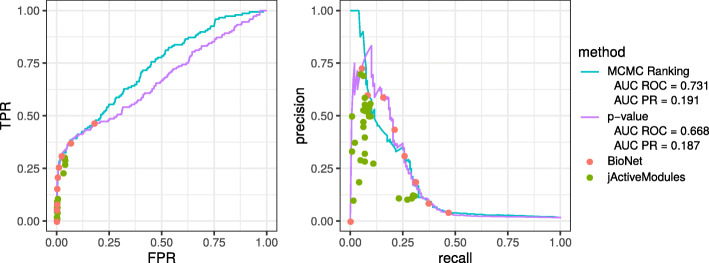
We propose a method to estimate probabilities of each vertex to belong to the active module based on Markov chain Monte Carlo (MCMC) subnetwork sampling. As an example of the performance of our method on real data, we run it on two gene expression datasets. For the first many-replicate expression dataset we show that the proposed approach is consistent with an existing resampling-based method. On the second dataset the jackknife resampling method is inapplicable due to the small number of biological replicates, but the MCMC method can be run and shows high classification performance.
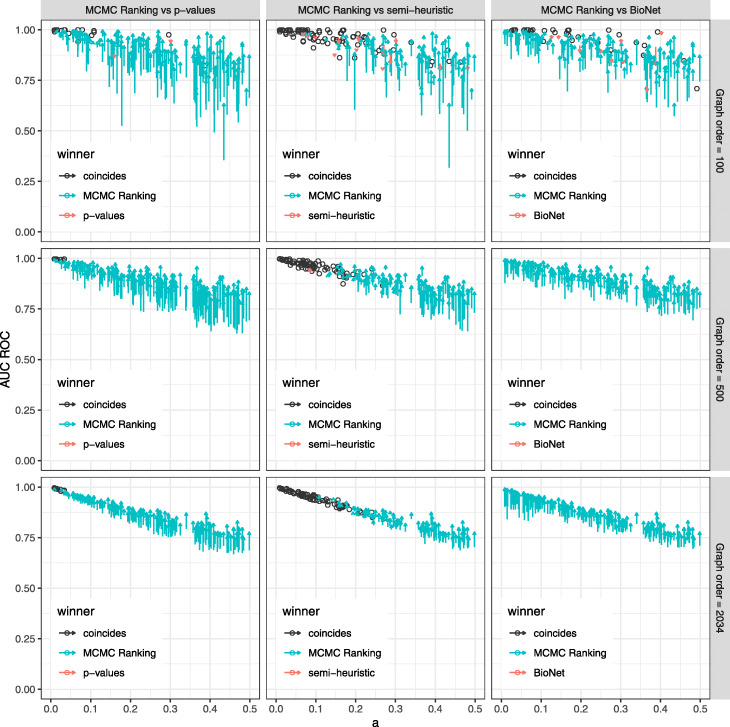
The proposed method allows to estimate the probability that an individual vertex belongs to the active module as well as the false discovery rate (FDR) for a given set of vertices. Given the estimated probabilities, it becomes possible to provide a connected subgraph in a consistent manner for any given FDR level: no vertex can disappear when the FDR level is relaxed. We show, on both simulated and real datasets, that the proposed method has good computational performance and high classification accuracy.
整合网络方法常用于解释高通量实验生物学数据:转录组学、蛋白质组学、代谢组学等。常见的方法之一是找到全局相互作用网络的一个连通子网络,该子网络最好包含数据中显著的个体变化,并代表所谓的活性模块。通常,实现此方法的方法会找到单个子网,从而为顶点解决硬分类问题。该子网本质上包含错误的顶点,而没有仪器可以估计任何特定顶点包含的置信度水平。为了解决这个问题,在当前的研究中,我们将活性模块问题视为软分类问题。
我们提出了一种基于马尔可夫链蒙特卡罗(MCMC)子网采样来估计每个顶点属于活性模块的概率的方法。作为我们的方法在真实数据上的性能的一个例子,我们在两个基因表达数据集上运行它。对于第一个具有许多重复表达的数据集,我们表明所提出的方法与基于现有重采样的方法一致。对于第二个数据集,由于生物学重复数较少,因此无法使用自举重采样方法,但可以运行 MCMC 方法,并且显示出较高的分类性能。
所提出的方法允许估计个体顶点属于活性模块的概率以及给定顶点集的错误发现率(FDR)。给定估计的概率,就可以以一致的方式为任何给定的 FDR 水平提供一个连通的子图:当 FDR 水平放宽时,没有顶点会消失。我们在模拟和真实数据集上都表明,所提出的方法具有良好的计算性能和高分类精度。