Noureddine Olfa, Issaoui Noureddine, Al-Dossary Omar
University of Monastir, Laboratory of Quantum and Statistical Physics (LR18ES18), Faculty of Sciences, Monastir 5079, Tunisia.
Department of Physics and Astronomy, College of Science, King Saud University, PO Box 2455, Riyadh 11451, Saudi Arabia.
J King Saud Univ Sci. 2021 Jan;33(1):101248. doi: 10.1016/j.jksus.2020.101248. Epub 2020 Nov 25.
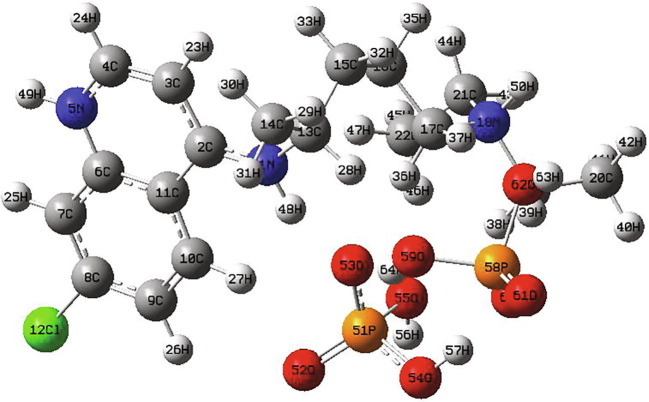
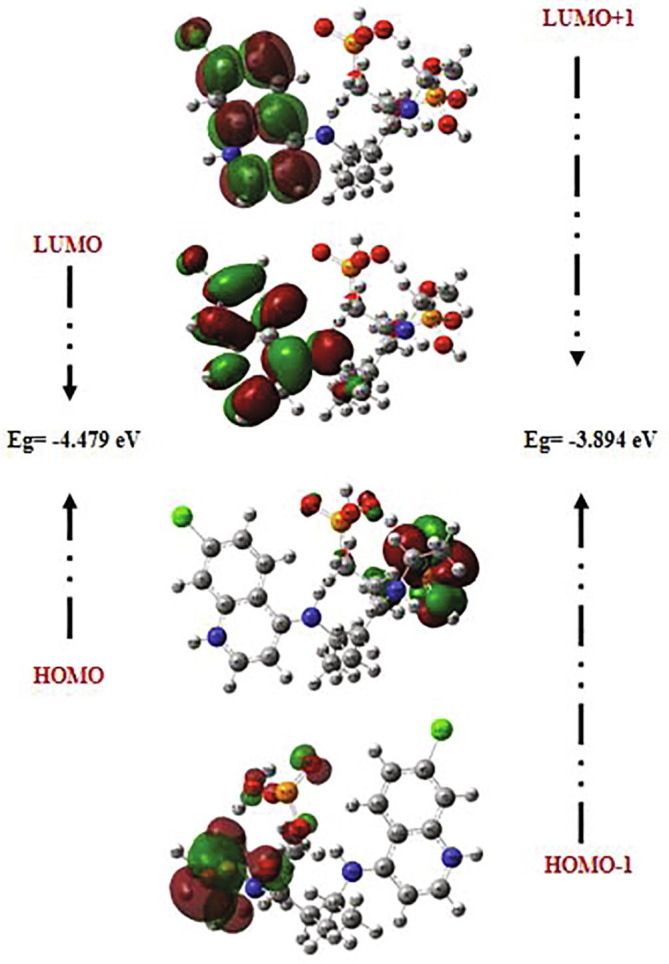
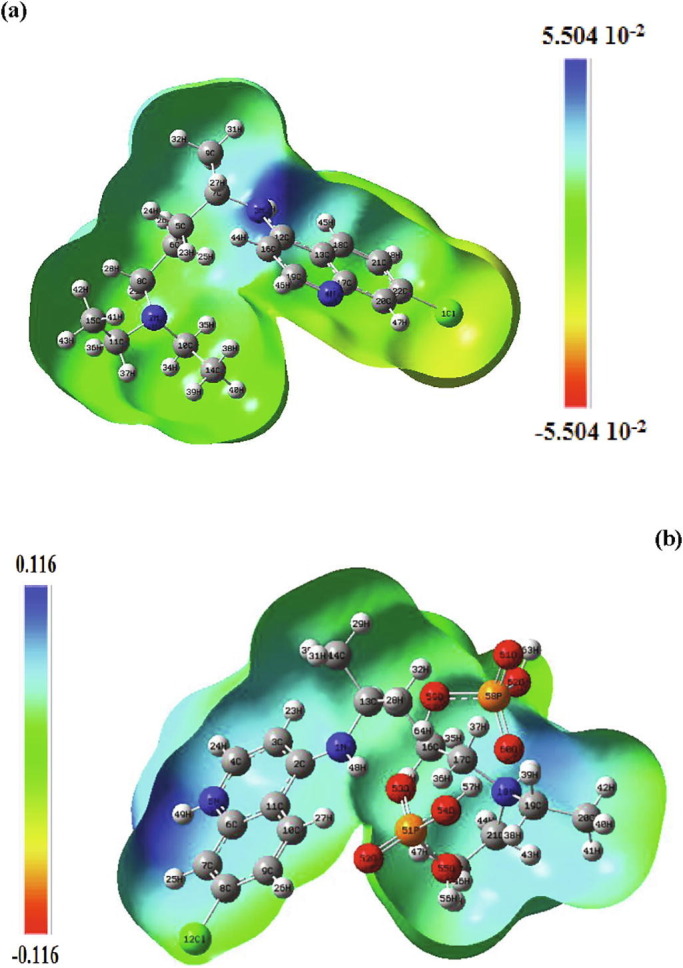
The recently emerged COVID-19 virus caused hundreds of thousands of deaths and instigated a widespread fear, threatening the world's most advanced health security. In 2020, chloroquine derivatives are among the drugs tested against the coronavirus pandemic and showed an apparent efficacy. In the present work, the chloroquine and the chloroquine phosphate molecules have been proposed as potential antiviral for the treatment of COVID-19 diseases combining DFT and molecular docking calculations. Molecular geometries, electronic properties and molecular electrostatic potential were investigated using density functional theory (DFT) at the B3LYP/6-31G* method. As results, we found a good agreement between the theoretical and the experimental geometrical parameters (bond lengths and bond angles). The frontier orbitals analysis has been calculated at the same level of theory to determine the charge transfer within the molecule. In order to perform a better description of the FMOs, the density of states was determined. The molecular electrostatic potential maps were calculated to provide information on the chemical reactivity of molecule and also to describe the intermolecular interactions. All these studies help us a lot in determining the reactivity of the mentioned compounds. Finally, docking calculations were carried out to determine the pharmaceutical activities of the chloroquine derivatives against coronavirus diseases. The choice of these ligands was based on their antiviral activities.
最近出现的新冠病毒导致数十万人死亡,并引发了广泛的恐惧,威胁到全球最先进的卫生安全。2020年,氯喹衍生物是针对新冠疫情进行测试的药物之一,并显示出明显的疗效。在本研究中,结合密度泛函理论(DFT)和分子对接计算,提出将氯喹和磷酸氯喹分子作为治疗新冠疾病的潜在抗病毒药物。使用B3LYP/6-31G*方法的密度泛函理论(DFT)研究了分子几何结构、电子性质和分子静电势。结果表明,理论几何参数(键长和键角)与实验值吻合良好。在相同理论水平下进行前沿轨道分析,以确定分子内的电荷转移。为了更好地描述前沿分子轨道,确定了态密度。计算分子静电势图,以提供有关分子化学反应性的信息,并描述分子间相互作用。所有这些研究对确定上述化合物的反应性有很大帮助。最后,进行对接计算以确定氯喹衍生物对冠状病毒疾病的药理活性。这些配体的选择基于它们的抗病毒活性。