Noureddine Olfa, Issaoui Noureddine, Medimagh Mouna, Al-Dossary Omar, Marouani Houda
University of Monastir, Laboratory of Quantum and Statistical Physics (LR18ES18), Faculty of Sciences, Monastir 5079, Tunisia.
Department of Physics and Astronomy, College of Science, King Saud University, PO Box 2455, Riyadh 11451, Saudi Arabia.
J King Saud Univ Sci. 2021 Mar;33(2):101334. doi: 10.1016/j.jksus.2020.101334. Epub 2021 Jan 6.
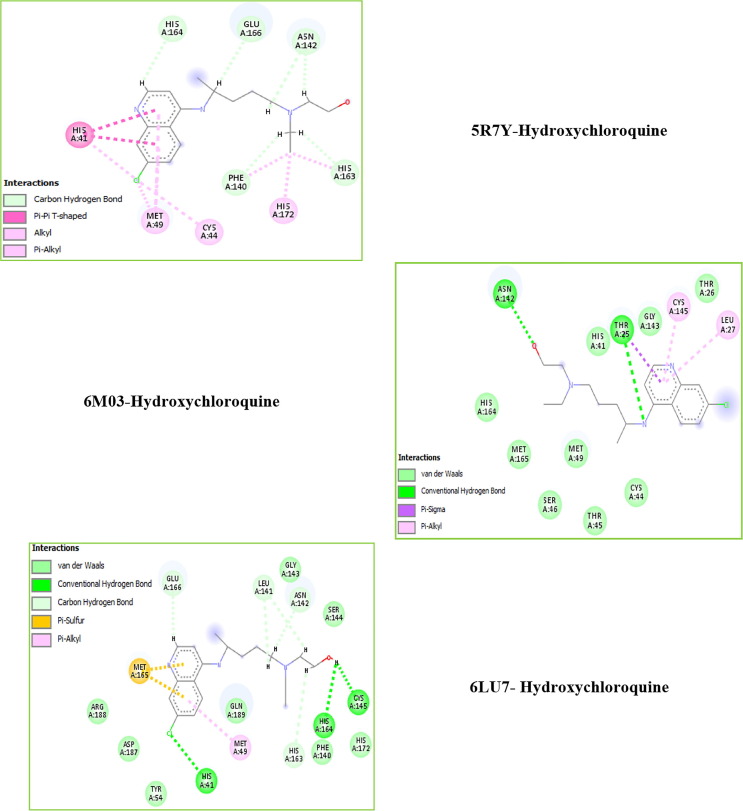
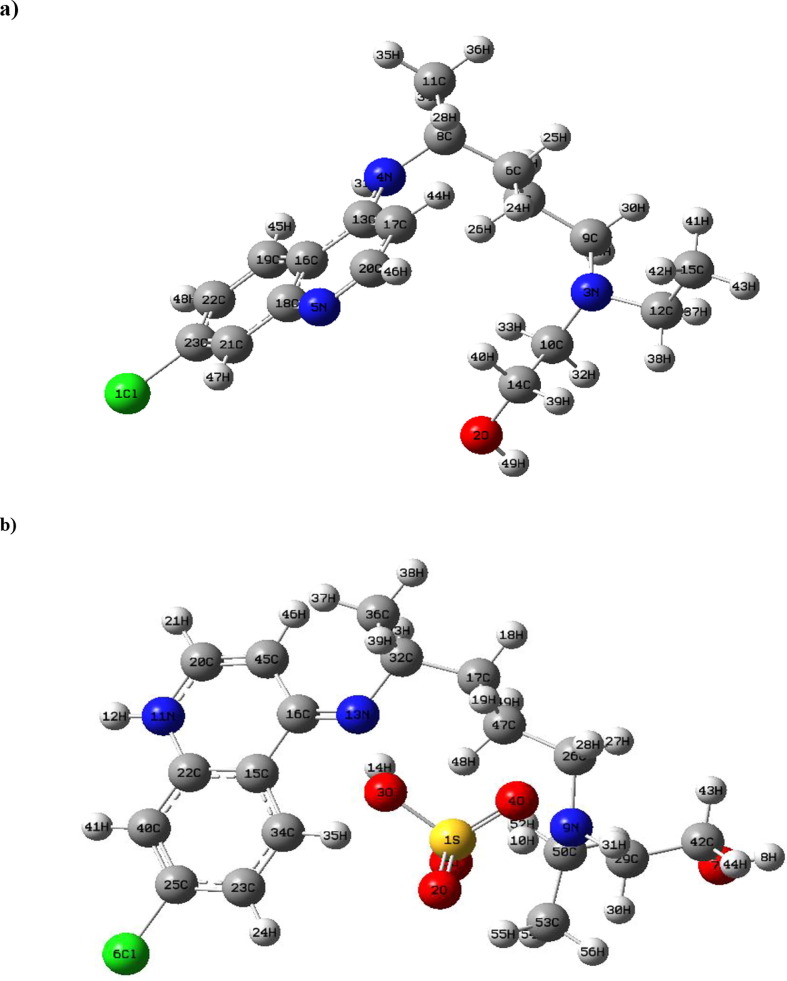
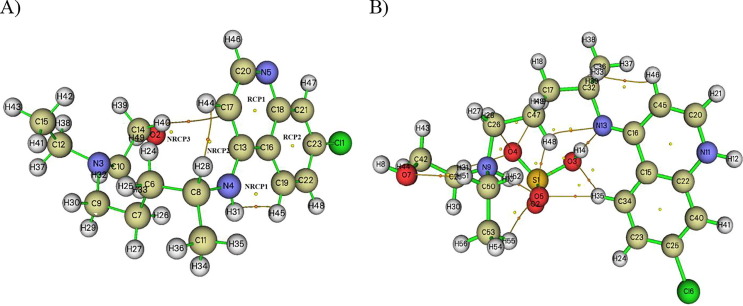
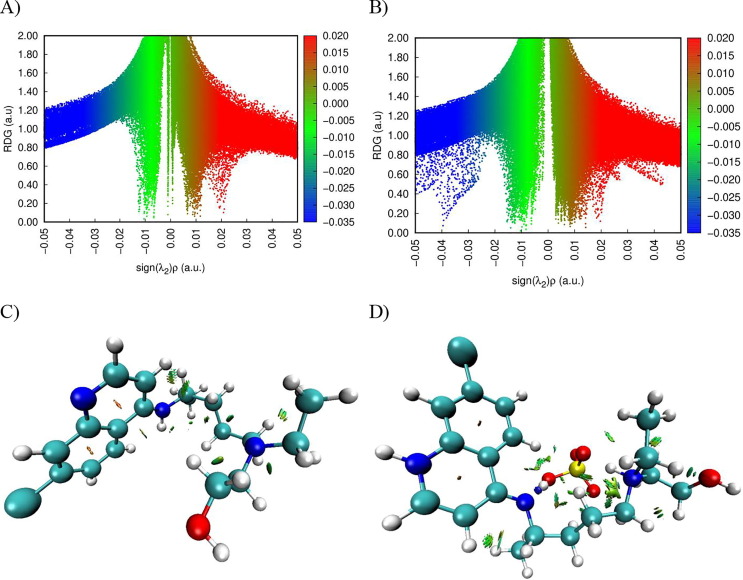
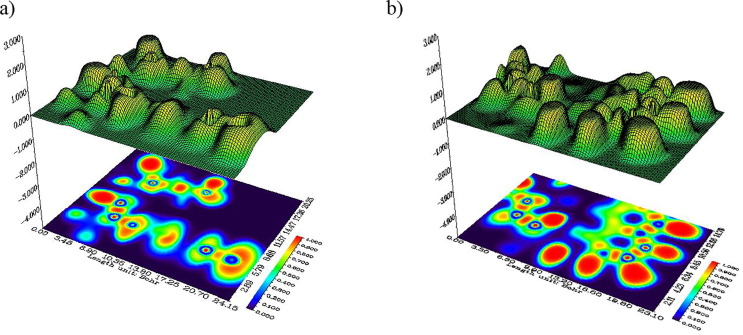
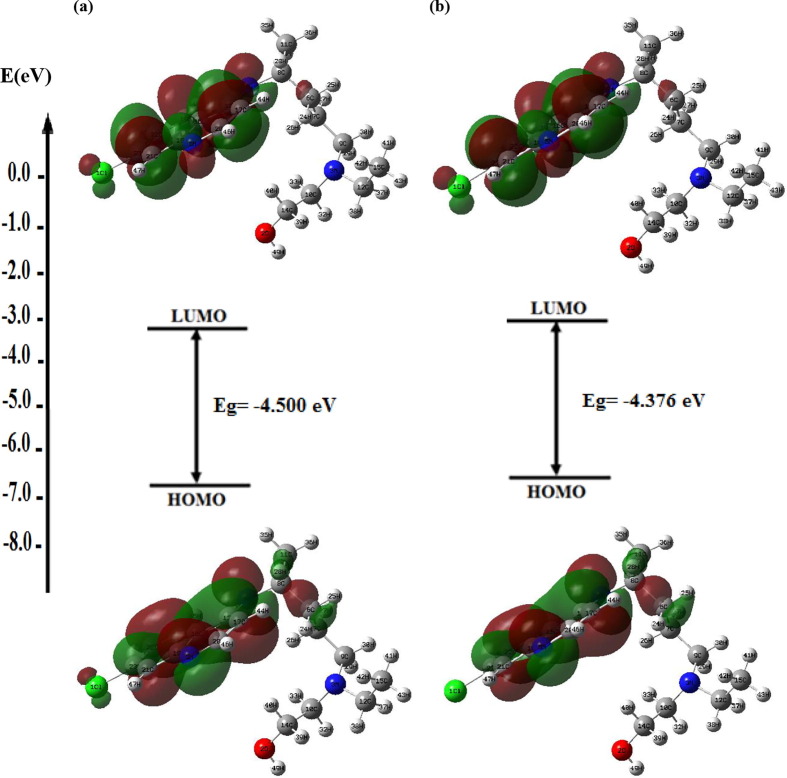
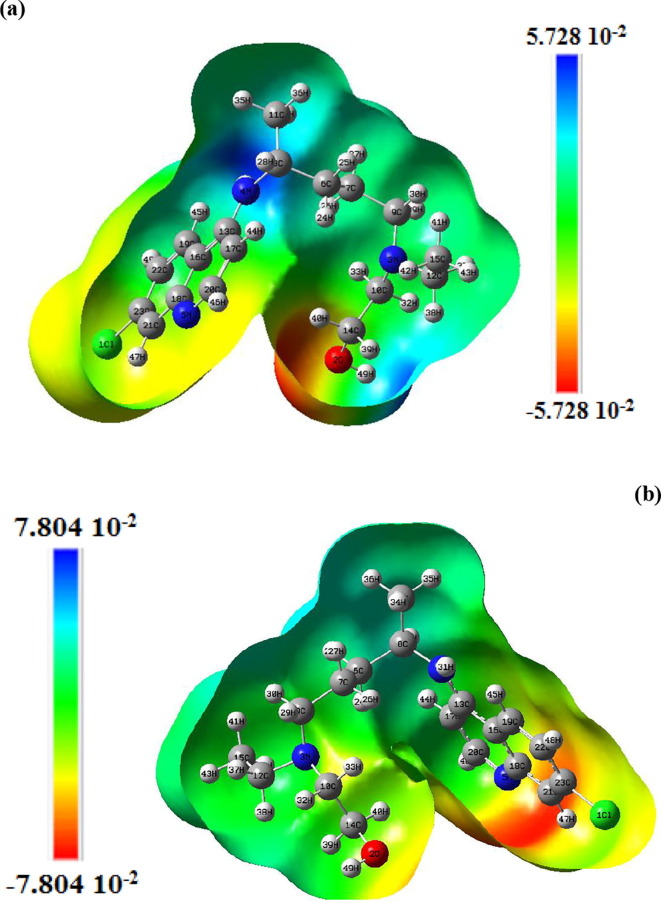
Structure-activity relationships for hydroxychloroquine compound and its derivatives resulted in a potent antiviral activity. Where hydroxychloroquine derivatives showed an apparent efficacy against coronavirus related pneumonia. For this reason, the current study is focused on the structural properties of hydroxychloroquine and hydroxychloroquine sulfate. Optimized structures of these molecules have been reported by using DFT method at B3LYP/6-31G* level of theory. The geometric were determined and compared with the experimental crystal structure. The intra and intermolecular interactions which exist within these compounds are analyzed by different methods namely the topological analysis AIM, ELF and the reduced gradient of the density. These approaches make it possible in particular to study the properties of hydrogen bonds. The highest occupied molecular orbital and the lowest unoccupied molecular orbital energy levels are constructed and the corresponding frontier energy gaps are determined to realize the charge transfer within the molecule. The densities of state diagrams were determined to calculate contributions to the molecular orbitals. The molecular electrostatic potential surfaces are determined to give a visual representation of charge distribution of these ligands and to provide information linked to electrophilic and nucleophilic sites localization. Finally, these derivatives were evaluated for the inhibition of COVID-19 activity by using the molecular docking method.
羟氯喹化合物及其衍生物的构效关系产生了强大的抗病毒活性。羟氯喹衍生物对冠状病毒相关肺炎显示出明显疗效。因此,当前研究聚焦于羟氯喹和硫酸羟氯喹的结构特性。已通过在B3LYP/6 - 31G*理论水平上使用密度泛函理论(DFT)方法报道了这些分子的优化结构。确定了几何结构并与实验晶体结构进行比较。通过不同方法,即拓扑分析AIM、电子定域函数(ELF)和密度的约化梯度,分析了这些化合物内部存在的分子内和分子间相互作用。这些方法尤其使得研究氢键的性质成为可能。构建了最高占据分子轨道和最低未占据分子轨道能级,并确定了相应的前沿能隙以实现分子内的电荷转移。确定了态密度图以计算对分子轨道的贡献。确定了分子静电势表面,以直观呈现这些配体的电荷分布,并提供与亲电和亲核位点定位相关的信息。最后,通过分子对接方法评估了这些衍生物对COVID - 19的抑制活性。