Department of Medical Microbiology, University Medical Center Utrecht, Utrecht, the Netherlands.
Department of Computer Science, Aalto University, FI-00076 Espoo, Finland.
Microb Genom. 2020 Dec;6(12). doi: 10.1099/mgen.0.000488. Epub 2020 Nov 30.
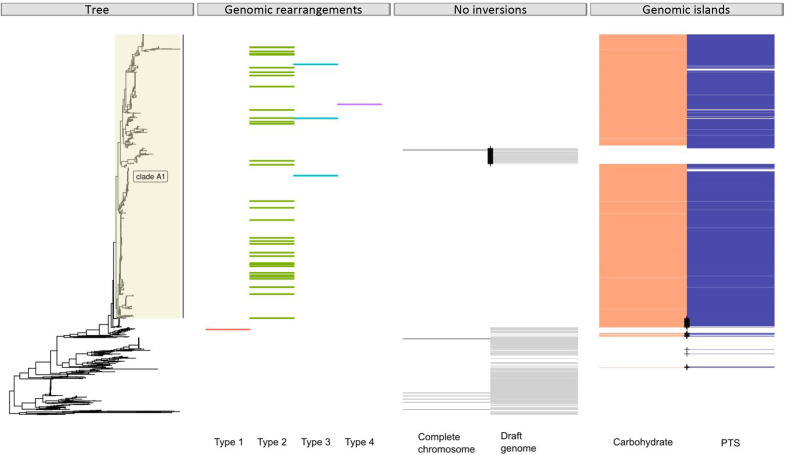
is a gut commensal of the gastro-digestive tract, but also known as nosocomial pathogen among hospitalized patients. Population genetics based on whole-genome sequencing has revealed that strains from hospitalized patients form a distinct clade, designated clade A1, and that plasmids are major contributors to the emergence of nosocomial . Here we further explored the adaptive evolution of using a genome-wide co-evolution study (GWES) to identify co-evolving single-nucleotide polymorphisms (SNPs). We identified three genomic regions harbouring large numbers of SNPs in tight linkage that are not proximal to each other based on the completely assembled chromosome of the clade A1 reference hospital isolate AUS0004. Close examination of these regions revealed that they are located at the borders of four different types of large-scale genomic rearrangements, insertion sites of two different genomic islands and an IS-like transposon. In non-clade A1 isolates, these regions are adjacent to each other and they lack the insertions of the genomic islands and IS-like transposon. Additionally, among the clade A1 isolates there is one group of pet isolates lacking the genomic rearrangement and insertion of the genomic islands, suggesting a distinct evolutionary trajectory. analysis of the biological functions of the genes encoded in three regions revealed a common link to a stress response. This suggests that these rearrangements may reflect adaptation to the stringent conditions in the hospital environment, such as antibiotics and detergents, to which bacteria are exposed. In conclusion, to our knowledge, this is the first study using GWES to identify genomic rearrangements, suggesting that there is considerable untapped potential to unravel hidden evolutionary signals from population genomic data.
是胃肠道的肠道共生菌,但也是住院患者中的医院病原体。基于全基因组测序的群体遗传学研究表明,来自住院患者的 菌株形成了一个独特的进化枝,称为 A1 进化枝,并且质粒是医院获得性 的主要贡献者。在这里,我们使用全基因组共进化研究(GWES)进一步探索了 的适应性进化,以鉴定共进化的单核苷酸多态性(SNP)。我们确定了三个含有大量紧密连锁 SNP 的基因组区域,这些 SNP 彼此之间不相邻,基于 A1 进化枝参考医院分离株 AUS0004 的完全组装染色体。对这些区域的仔细检查表明,它们位于四个不同类型的大规模基因组重排、两个不同基因组岛的插入位点和一个 IS 样转座子的边界。在非 A1 进化枝分离株中,这些区域彼此相邻,并且缺乏基因组岛和 IS 样转座子的插入。此外,在 A1 进化枝分离株中,有一组宠物分离株缺乏基因组重排和基因组岛的插入,表明存在明显不同的进化轨迹。对三个区域编码基因的生物学功能进行 分析表明,它们与应激反应有共同的联系。这表明这些重排可能反映了对医院环境中严格条件的适应,例如细菌暴露于抗生素和清洁剂。总之,据我们所知,这是首次使用 GWES 鉴定基因组重排的研究,表明从群体基因组数据中揭示隐藏的进化信号还有很大的潜力尚未挖掘。