Vanderbilt University, Department of Biological Sciences, Nashville, Tennessee, United States of America.
Nashville, Tennessee, United States of America.
PLoS Biol. 2020 Dec 2;18(12):e3001007. doi: 10.1371/journal.pbio.3001007. eCollection 2020 Dec.
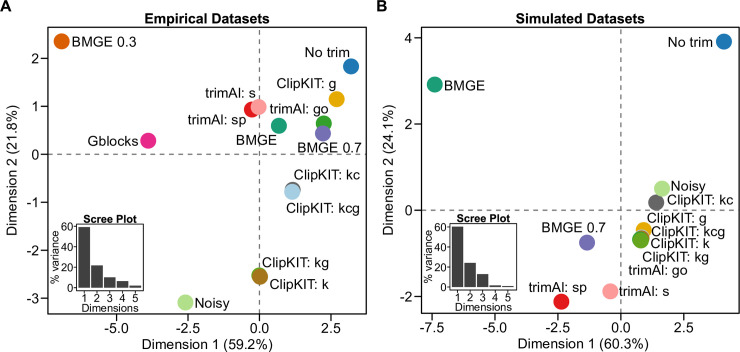
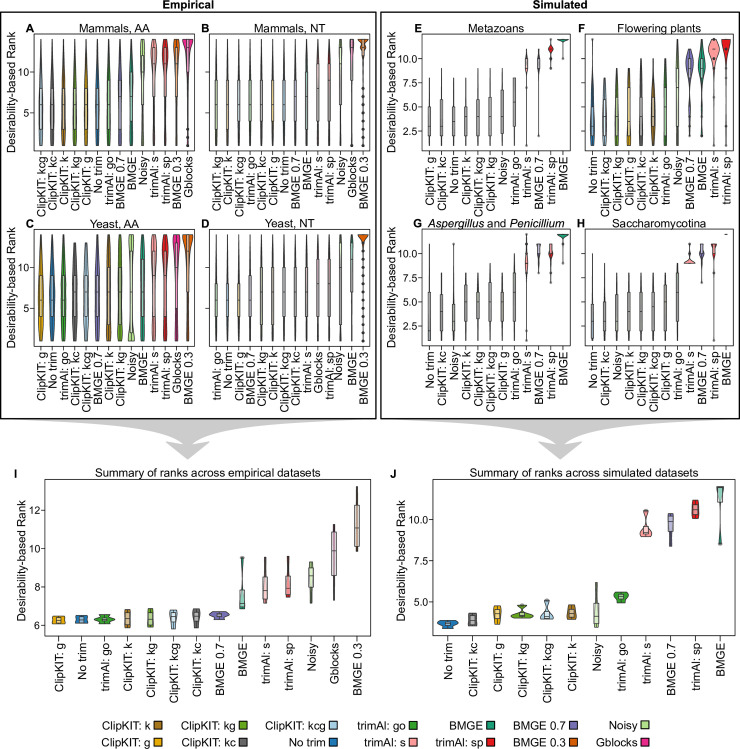
Highly divergent sites in multiple sequence alignments (MSAs), which can stem from erroneous inference of homology and saturation of substitutions, are thought to negatively impact phylogenetic inference. Thus, several different trimming strategies have been developed for identifying and removing these sites prior to phylogenetic inference. However, a recent study reported that doing so can worsen inference, underscoring the need for alternative alignment trimming strategies. Here, we introduce ClipKIT, an alignment trimming software that, rather than identifying and removing putatively phylogenetically uninformative sites, instead aims to identify and retain parsimony-informative sites, which are known to be phylogenetically informative. To test the efficacy of ClipKIT, we examined the accuracy and support of phylogenies inferred from 14 different alignment trimming strategies, including those implemented in ClipKIT, across nearly 140,000 alignments from a broad sampling of evolutionary histories. Phylogenies inferred from ClipKIT-trimmed alignments are accurate, robust, and time saving. Furthermore, ClipKIT consistently outperformed other trimming methods across diverse datasets, suggesting that strategies based on identifying and retaining parsimony-informative sites provide a robust framework for alignment trimming.
高度分化的多序列比对(MSA)位点可能源于同源性推断错误和替代饱和,被认为会对系统发育推断产生负面影响。因此,已经开发了几种不同的修剪策略,用于在进行系统发育推断之前识别和去除这些位点。然而,最近的一项研究报告称,这样做可能会恶化推断,这凸显了需要替代的对齐修剪策略。在这里,我们介绍了 ClipKIT,这是一种对齐修剪软件,它不是识别和去除推测的系统发育上无信息的位点,而是旨在识别和保留简约信息位点,这些位点已知是系统发育信息丰富的。为了测试 ClipKIT 的功效,我们检查了来自 ClipKIT 等 14 种不同对齐修剪策略推断的系统发育的准确性和支持率,这些策略涵盖了来自广泛进化历史样本的近 140,000 个对齐。从 ClipKIT 修剪对齐推断出的系统发育是准确的、稳健的且节省时间的。此外,ClipKIT 在不同数据集上始终优于其他修剪方法,这表明基于识别和保留简约信息位点的策略为对齐修剪提供了一个稳健的框架。