Hassan Wafaa M M, Tawab Ashraf A Abd El, El-Shannat Sara M
Reference Laboratory for Quality Control on Poultry Production, Animal Health Research Institute, Dokki, Giza, Egypt.
Department of Bacteriology, Immunology, and Mycology, Faculty of Veterinary Medicine, Benha University, Benha, Egypt.
Vet World. 2020 Oct;13(10):2252-2259. doi: 10.14202/vetworld.2020.2252-2259. Epub 2020 Oct 28.
This study aimed to characterize the genetic diversity, evolutionary level, and prevalence of genotypes of common isolates of ( Enteritidis and Typhimurium). Using one of the most advanced molecular recognition techniques, multilocus variable number of tandem repeat analysis (MLVA), we characterized the genotype and prevalence of . Enteritidis and . Typhimurium.
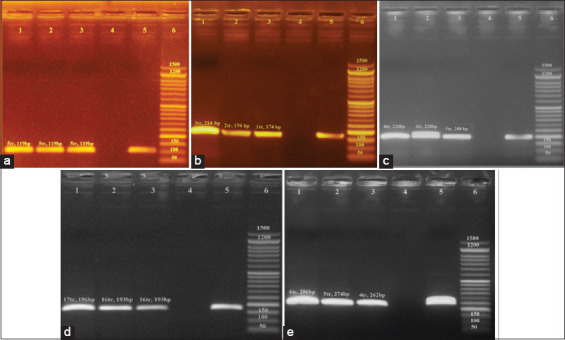
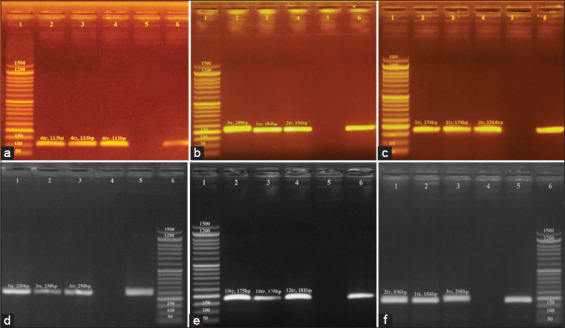
One hundred and twenty-five internal organ samples were collected from the major chicken slaughterhouses in Egypt, and species were isolated. PCR was utilized to amplify the and genes to identify . Enteritidis and . Typhimurium DNA, respectively, from isolates. MLVA was applied on nine samples of . Enteritidis DNA and three samples of . Typhimurium DNA. Six variable number tandem repeat (VNTR) loci (Sal02, Sal04, Sal06, Sal10, Sal20, and Sal23) were amplified.
Of the examined samples (n=125), a total of 12 isolates (9.6%) were either identified as Enteritidis or Typhimurium. PCR-mediated amplification of and revealed that 75% (n=9) of the 12 isolates were . Enteritidis and 25% (n=3) were . Typhimurium. The six loci amplified through MLVA had allelic diversity. The most discriminatory heterogenic locus for . Enteritidis was Sal20. Sal04 and Sal23 were the most discriminatory heterogenic loci for . Typhimurium. VNTR allelic profile analysis revealed nine unique genotypes for . Enteritidis and three for . Typhimurium.
This study was the first to use MLVA analysis to identify . Enteritidis and . Typhimurium strains isolated from chickens in Egypt. The molecular typing data reported herein allowed us to characterize the genotypes of . Enteritidis and . Typhimurium that are most prevalent in Egyptian chickens. Moreover, this epidemiological information provides valuable insight on how to prevent disease transmission. Moreover, our methods provide an alternative to traditional serotyping techniques that may produce inaccurate strain identifications for organisms with rough lipopolysaccharide structures.
本研究旨在对肠炎沙门氏菌和鼠伤寒沙门氏菌常见分离株的遗传多样性、进化水平及基因型流行情况进行特征分析。我们使用最先进的分子识别技术之一,多位点可变数目串联重复序列分析(MLVA),对肠炎沙门氏菌和鼠伤寒沙门氏菌的基因型及流行情况进行了特征分析。
从埃及主要的鸡肉屠宰场收集了125份内脏样本,并分离出沙门氏菌属菌种。利用聚合酶链反应(PCR)扩增invA和stn基因,分别从分离株中鉴定肠炎沙门氏菌和鼠伤寒沙门氏菌的DNA。对9份肠炎沙门氏菌DNA样本和3份鼠伤寒沙门氏菌DNA样本进行MLVA分析。扩增了6个可变数目串联重复序列(VNTR)位点(Sal02、Sal04、Sal06、Sal10、Sal20和Sal23)。
在所检测的125份样本中,共有12株分离株(9.6%)被鉴定为肠炎沙门氏菌或鼠伤寒沙门氏菌。PCR介导的invA和stn扩增显示,12株沙门氏菌分离株中有75%(n = 9)为肠炎沙门氏菌,25%(n = 3)为鼠伤寒沙门氏菌。通过MLVA扩增的6个位点具有等位基因多样性。肠炎沙门氏菌最具鉴别力的异质性位点是Sal20。Sal04和Sal23是鼠伤寒沙门氏菌最具鉴别力的异质性位点。VNTR等位基因谱分析显示肠炎沙门氏菌有9种独特基因型,鼠伤寒沙门氏菌有3种。
本研究首次使用MLVA分析来鉴定从埃及鸡中分离出的肠炎沙门氏菌和鼠伤寒沙门氏菌菌株。本文报道的分子分型数据使我们能够对埃及鸡中最常见的肠炎沙门氏菌和鼠伤寒沙门氏菌基因型进行特征分析。此外,这些流行病学信息为如何预防疾病传播提供了有价值的见解。而且,我们的方法为传统血清分型技术提供了一种替代方法,传统血清分型技术可能会对具有粗糙脂多糖结构的生物体产生不准确的菌株鉴定。