MS Proteomics Research Group, Research Centre for Natural Sciences, Magyar Tudósok Körútja 2, H-1117 Budapest, Hungary.
Faculty of Science, Institute of Chemistry, Hevesy György PhD School of Chemistry, ELTE, Eötvös Loránd University, Pázmány Péter Sétány 1/A, H-1117 Budapest, Hungary.
J Proteome Res. 2021 Jan 1;20(1):474-484. doi: 10.1021/acs.jproteome.0c00518. Epub 2020 Dec 7.
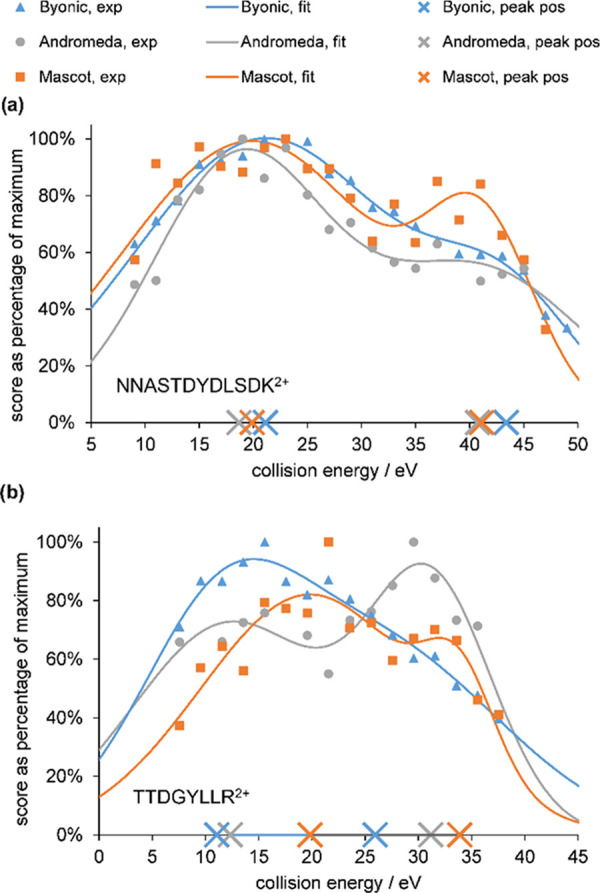
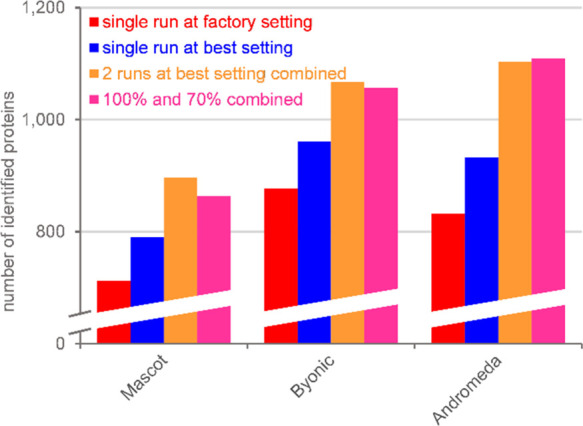
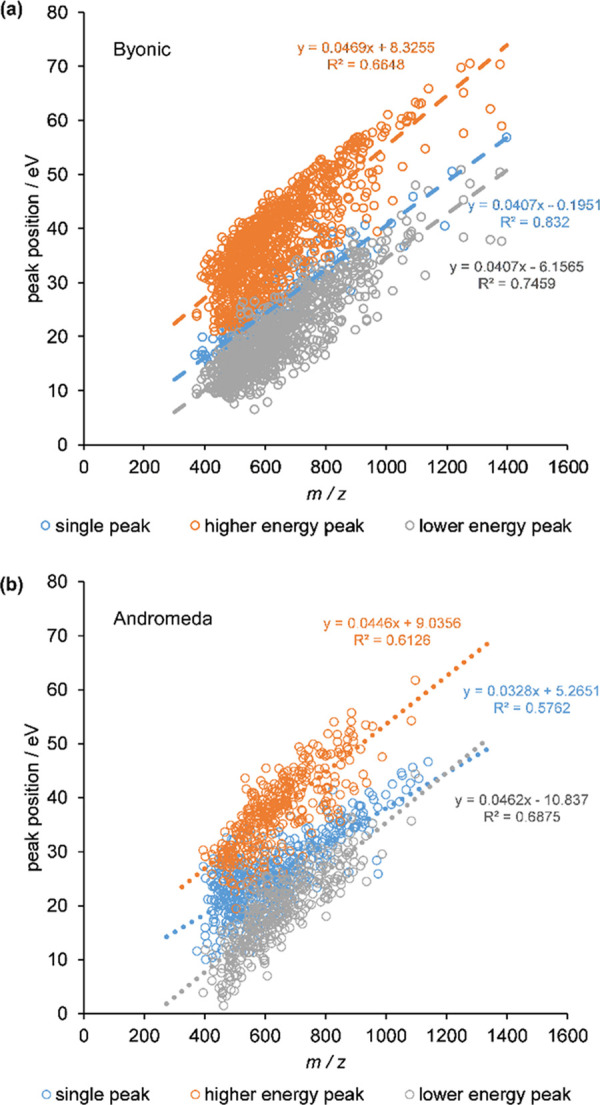
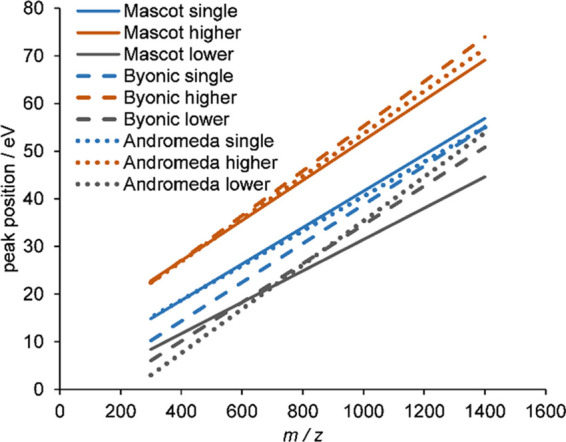
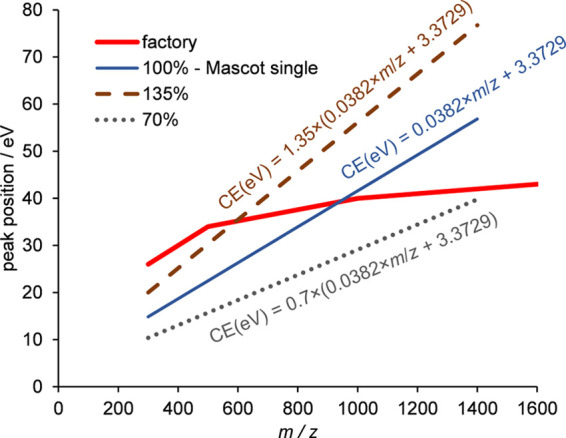
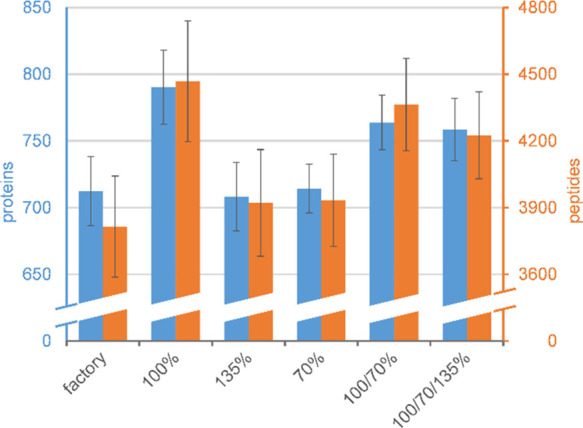
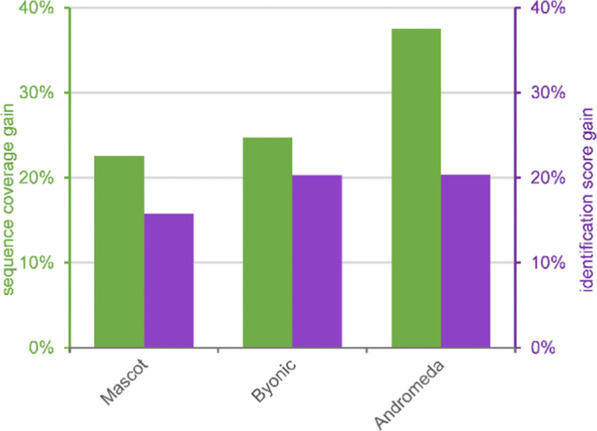
Bottom-up proteomics relies on identification of peptides from tandem mass spectra, usually via matching against sequence databases. Confidence in a peptide-spectrum match can be characterized by a score value given by the database search engines, and it depends on the information content and the quality of the spectrum. The latter are influenced by experimental parameters, of which the collision energy is the most important one in the case of collision-induced dissociation. We examined how the identification score of the Byonic and Andromeda (MaxQuant) engines varies with collision energy for more than a thousand individual peptides from a HeLa tryptic digest on a QTof instrument. We thereby extended our earlier study on Mascot scores and corroborated its findings on the potential bimodal nature of this energy dependence. Optimal energies as a function of / show comparable linear trends for the three engines. On the basis of peptide-level results, we designed methods with one or two liquid chromatography-tandem mass spectrometry (LC-MS/MS) runs and various collision energy settings and assessed their practical performance in peptide and protein identification from the HeLa standard sample. A 10-40% gain in various measures, such as the number of identified proteins or sequence coverage, was obtained over the factory default settings. Best performing methods differ for the three engines, suggesting that the experimental parameters should be fine-tuned to the choice of the engine. We also recommend a simple approach and provide reference data to ease the transfer of the optimized methods to other mass spectrometers relevant for proteomics. We demonstrate the utility of this approach on an Orbitrap instrument. Data sets can be accessed via the MassIVE repository (MSV000086379).
自下而上的蛋白质组学依赖于通过串联质谱的肽的鉴定,通常是通过与序列数据库匹配。肽谱匹配的置信度可以通过数据库搜索引擎给出的评分值来描述,该值取决于信息含量和光谱的质量。后者受实验参数的影响,在碰撞诱导解离的情况下,碰撞能是最重要的。我们检查了在 QTof 仪器上对 HeLa 酶解物进行的超过一千个单个肽的情况下,Byonic 和 Andromeda(MaxQuant)引擎的鉴定得分如何随碰撞能而变化。我们扩展了之前关于 Mascot 分数的研究,并证实了其关于这种能量依赖性的潜在双峰性质的发现。作为 / 的函数的最佳能量对于三个引擎具有可比的线性趋势。基于肽级别的结果,我们设计了具有一个或两个液相色谱-串联质谱(LC-MS/MS)运行和各种碰撞能设置的方法,并评估了它们在从 HeLa 标准样品中进行肽和蛋白质鉴定方面的实际性能。与工厂默认设置相比,各种措施(例如鉴定的蛋白质数量或序列覆盖率)提高了 10-40%。对于三个引擎,表现最佳的方法不同,这表明应根据引擎的选择对实验参数进行微调。我们还建议一种简单的方法,并提供参考数据,以方便将优化的方法转移到其他与蛋白质组学相关的质谱仪上。我们在 Orbitrap 仪器上证明了这种方法的实用性。数据集可以通过 MassIVE 存储库(MSV000086379)访问。