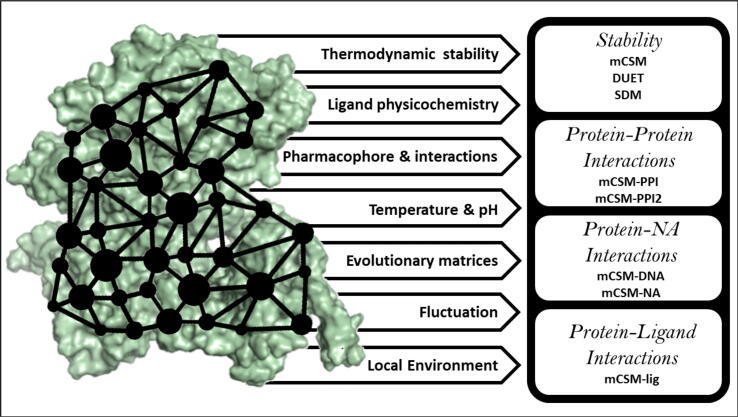
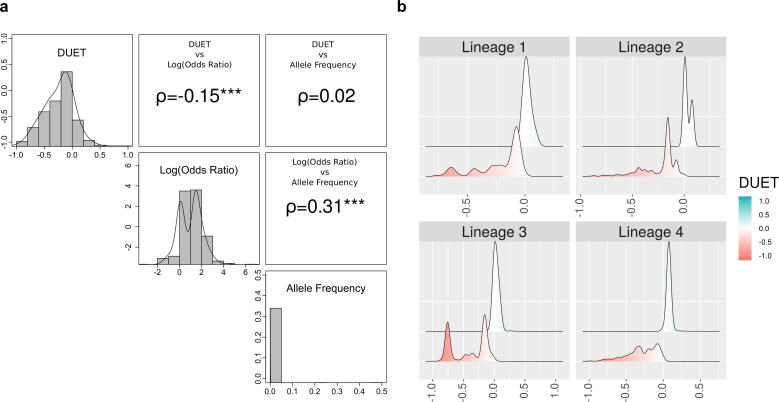
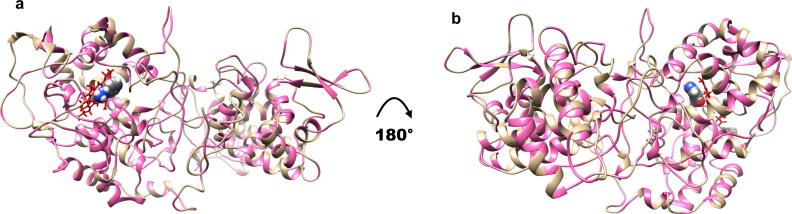
Tunstall Tanushree, Portelli Stephanie, Phelan Jody, Clark Taane G, Ascher David B, Furnham Nicholas
Department of Infection Biology, London School of Hygiene and Tropical Medicine, Keppel Street, London WC1E 7HT, UK.
Computational Biology and Clinical Informatics, Baker Heart and Diabetes Institute, Australia.
Comput Struct Biotechnol J. 2020 Oct 29;18:3377-3394. doi: 10.1016/j.csbj.2020.10.017. eCollection 2020.
Antimicrobials against bacterial, viral and parasitic pathogens have transformed human and animal health. Nevertheless, their widespread use (and misuse) has led to the emergence of antimicrobial resistance (AMR) which poses a potentially catastrophic threat to public health and animal husbandry. There are several routes, both intrinsic and acquired, by which AMR can develop. One major route is through non-synonymous single nucleotide polymorphisms (nsSNPs) in coding regions. Large scale genomic studies using high-throughput sequencing data have provided powerful new ways to rapidly detect and respond to such genetic mutations linked to AMR. However, these studies are limited in their mechanistic insight. Computational tools can rapidly and inexpensively evaluate the effect of mutations on protein function and evolution. Subsequent insights can then inform experimental studies, and direct existing or new computational methods. Here we review a range of sequence and structure-based computational tools, focussing on tools successfully used to investigate mutational effect on drug targets in clinically important pathogens, particularly . Combining genomic results with the biophysical effects of mutations can help reveal the molecular basis and consequences of resistance development. Furthermore, we summarise how the application of such a mechanistic understanding of drug resistance can be applied to limit the impact of AMR.
针对细菌、病毒和寄生虫病原体的抗菌药物改变了人类和动物的健康状况。然而,它们的广泛使用(以及滥用)导致了抗菌药物耐药性(AMR)的出现,这对公共卫生和畜牧业构成了潜在的灾难性威胁。AMR的产生有多种途径,包括内在途径和获得途径。一个主要途径是通过编码区的非同义单核苷酸多态性(nsSNPs)。利用高通量测序数据进行的大规模基因组研究提供了强大的新方法,能够快速检测并应对与AMR相关的此类基因突变。然而,这些研究在机理洞察方面存在局限性。计算工具可以快速且低成本地评估突变对蛋白质功能和进化的影响。随后的洞察结果可为实验研究提供参考,并指导现有的或新的计算方法。在此,我们综述了一系列基于序列和结构的计算工具,重点关注成功用于研究临床上重要病原体中突变对药物靶点影响的工具,特别是 。将基因组结果与突变的生物物理效应相结合,有助于揭示耐药性产生的分子基础和后果。此外,我们总结了如何应用这种对耐药性的机理理解来限制AMR的影响。