Structural Bioinformatics Lab, Department of Experimental and Health Science, Universitat Pompeu Fabra, 08003, Barcelona, Catalonia, Spain.
Laboratory of Protein Design and Immuno-Enginneering, School of Engineering, Ecole Polytechnique Federale de Lausanne, 1015, Lausanne, Vaud, Switzerland.
BMC Bioinformatics. 2021 Jan 6;22(1):4. doi: 10.1186/s12859-020-03770-5.
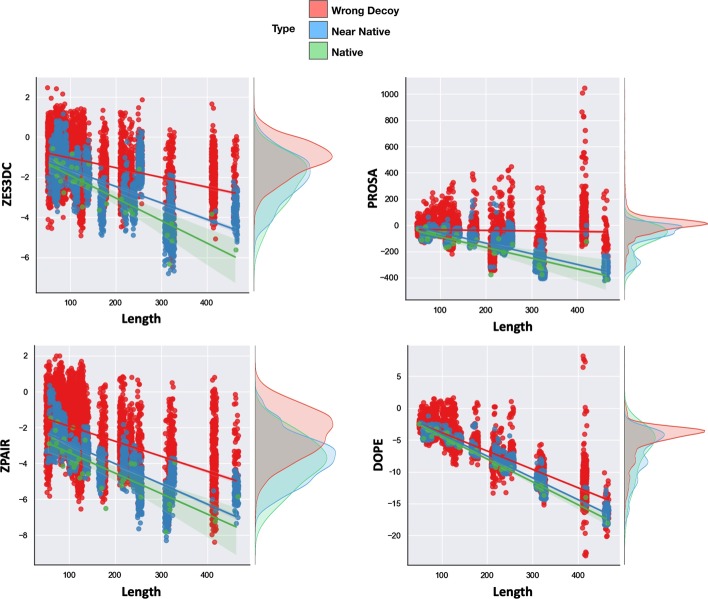
Statistical potentials, also named knowledge-based potentials, are scoring functions derived from empirical data that can be used to evaluate the quality of protein folds and protein-protein interaction (PPI) structures. In previous works we decomposed the statistical potentials in different terms, named Split-Statistical Potentials, accounting for the type of amino acid pairs, their hydrophobicity, solvent accessibility and type of secondary structure. These potentials have been successfully used to identify near-native structures in protein structure prediction, rank protein docking poses, and predict PPI binding affinities.
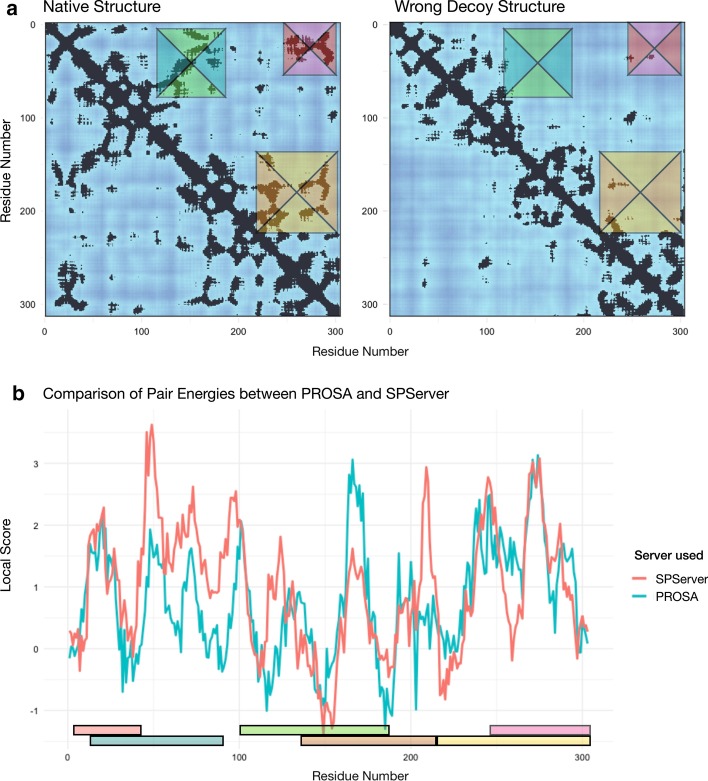
Here, we present the SPServer, a web server that applies the Split-Statistical Potentials to analyze protein folds and protein interfaces. SPServer provides global scores as well as residue/residue-pair profiles presented as score plots and maps. This level of detail allows users to: (1) identify potentially problematic regions on protein structures; (2) identify disrupting amino acid pairs in protein interfaces; and (3) compare and analyze the quality of tertiary and quaternary structural models.
While there are many web servers that provide scoring functions to assess the quality of either protein folds or PPI structures, SPServer integrates both aspects in a unique easy-to-use web server. Moreover, the server permits to locally assess the quality of the structures and interfaces at a residue level and provides tools to compare the local assessment between structures. SERVER ADDRESS: https://sbi.upf.edu/spserver/ .
统计势,也称为基于知识的势,是从经验数据中得出的评分函数,可用于评估蛋白质折叠和蛋白质-蛋白质相互作用(PPI)结构的质量。在以前的工作中,我们将统计势分解为不同的项,称为 Split-Statistical Potentials,分别考虑氨基酸对的类型、疏水性、溶剂可及性和二级结构的类型。这些势已成功用于识别蛋白质结构预测中的近天然结构、对蛋白质对接构象进行排序以及预测 PPI 结合亲和力。
在这里,我们展示了 SPServer,这是一个应用 Split-Statistical Potentials 来分析蛋白质折叠和蛋白质界面的网络服务器。SPServer 提供全局评分以及作为评分图和映射呈现的残基/残基对分布。这种详细程度允许用户:(1)识别蛋白质结构上可能存在问题的区域;(2)识别蛋白质界面中破坏的氨基酸对;(3)比较和分析三级和四级结构模型的质量。
虽然有许多网络服务器提供评分函数来评估蛋白质折叠或 PPI 结构的质量,但 SPServer 将这两个方面集成在一个独特的易于使用的网络服务器中。此外,该服务器允许在残基水平上局部评估结构和界面的质量,并提供工具来比较结构之间的局部评估。服务器地址:https://sbi.upf.edu/spserver/。