McNair Katelyn, Ecale Zhou Carol L, Souza Brian, Malfatti Stephanie, Edwards Robert A
Computational Sciences Research Center, San Diego State University, 5500 Campanile Drive, San Diego, CA 92182, USA.
Lawrence Livermore National Laboratory, Global Security Computing Applications, Livermore, CA 94550, USA.
Microorganisms. 2021 Jan 8;9(1):129. doi: 10.3390/microorganisms9010129.
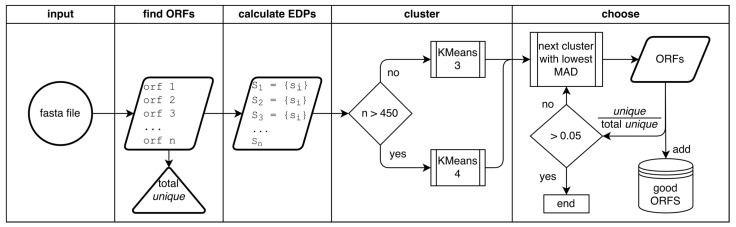
One of the main steps in gene-finding in prokaryotes is determining which open reading frames encode for a protein, and which occur by chance alone. There are many different methods to differentiate the two; the most prevalent approach is using shared homology with a database of known genes. This method presents many pitfalls, most notably the catch that you only find genes that you have seen before. The four most popular prokaryotic gene-prediction programs (GeneMark, Glimmer, Prodigal, Phanotate) all use a protein-coding training model to predict protein-coding genes, with the latter three allowing for the training model to be created ab initio from the input genome. Different methods are available for creating the training model, and to increase the accuracy of such tools, we present here GOODORFS, a method for identifying protein-coding genes within a set of all possible open reading frames (ORFS). Our workflow begins with taking the amino acid frequencies of each ORF, calculating an entropy density profile (EDP), using KMeans to cluster the EDPs, and then selecting the cluster with the lowest variation as the coding ORFs. To test the efficacy of our method, we ran GOODORFS on 14,179 annotated phage genomes, and compared our results to the initial training-set creation step of four other similar methods (Glimmer, MED2, PHANOTATE, Prodigal). We found that GOODORFS was the most accurate (0.94) and had the best F1-score (0.85), while Glimmer had the highest precision (0.92) and PHANOTATE had the highest recall (0.96).
原核生物基因寻找中的主要步骤之一是确定哪些开放阅读框编码蛋白质,哪些只是偶然出现。有许多不同的方法来区分这两者;最普遍的方法是与已知基因数据库进行共享同源性比较。这种方法存在许多缺陷,最明显的是只能找到之前见过的基因。四个最流行的原核生物基因预测程序(GeneMark、Glimmer、Prodigal、Phanotate)都使用蛋白质编码训练模型来预测蛋白质编码基因,后三个程序允许从输入基因组从头创建训练模型。有不同的方法可用于创建训练模型,为了提高此类工具的准确性,我们在此介绍GOODORFS,一种在所有可能的开放阅读框(ORF)集合中识别蛋白质编码基因的方法。我们的工作流程首先获取每个ORF的氨基酸频率,计算熵密度分布(EDP),使用KMeans对EDP进行聚类,然后选择变异最小的聚类作为编码ORF。为了测试我们方法的有效性,我们在14179个注释的噬菌体基因组上运行了GOODORFS,并将我们的结果与其他四种类似方法(Glimmer、MED2、PHANOTATE、Prodigal)的初始训练集创建步骤进行了比较。我们发现GOODORFS最准确(0.94)且F1分数最高(0.85),而Glimmer精度最高(0.92),PHANOTATE召回率最高(0.96)。