Lee Sangkyu, Deasy Joseph O, Oh Jung Hun, Di Meglio Antonio, Dumas Agnes, Menvielle Gwenn, Charles Cecile, Boyault Sandrine, Rousseau Marina, Besse Celine, Thomas Emilie, Boland Anne, Cottu Paul, Tredan Olivier, Levy Christelle, Martin Anne-Laure, Everhard Sibille, Ganz Patricia A, Partridge Ann H, Michiels Stefan, Deleuze Jean-François, Andre Fabrice, Vaz-Luis Ines
Memorial Sloan Kettering Cancer Center, New York, NY, USA.
Gustave Roussy, INSERM Unit 981, Villejuif, France.
JNCI Cancer Spectr. 2020 May 11;4(5):pkaa039. doi: 10.1093/jncics/pkaa039. eCollection 2020 Oct.
We aimed at predicting fatigue after breast cancer treatment using machine learning on clinical covariates and germline genome-wide data.
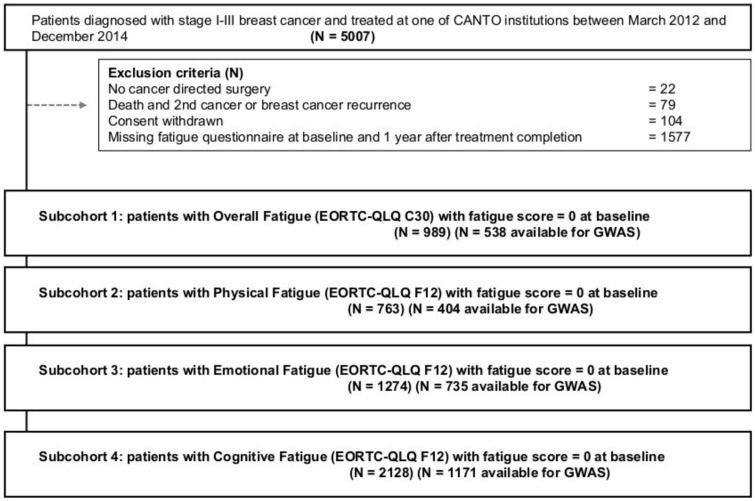
We accessed germline genome-wide data of 2799 early-stage breast cancer patients from the Cancer Toxicity study (NCT01993498). The primary endpoint was defined as scoring zero at diagnosis and higher than quartile 3 at 1 year after primary treatment completion on European Organization for Research and Treatment of Cancer quality-of-life questionnaires for Overall Fatigue and on the multidimensional questionnaire for Physical, Emotional, and Cognitive fatigue. First, we tested univariate associations of each endpoint with clinical variables and genome-wide variants. Then, using preselected clinical (false discovery rate < 0.05) and genomic ( < .001) variables, a multivariable preconditioned random-forest regression model was built and validated on a hold-out subset to predict fatigue. Gene set enrichment analysis identified key biological correlates (MetaCore). All statistical tests were 2-sided.
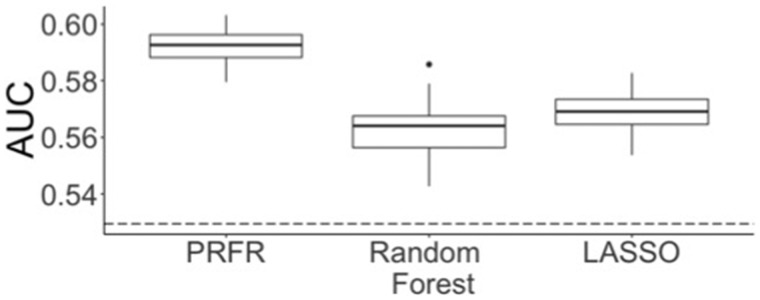
Statistically significant clinical associations were found only with Emotional and Cognitive Fatigue, including receipt of chemotherapy, anxiety, and pain. Some single nucleotide polymorphisms had some degree of association ( < .001) with the different fatigue endpoints, although there were no genome-wide statistically significant ( < 5.00 × 10) associations. Only for Cognitive Fatigue, the predictive ability of the genomic multivariable model was statistically significantly better than random (area under the curve = 0.59, = .01) and marginally improved with clinical variables (area under the curve = 0.60, = .005). Single nucleotide polymorphisms found to be associated ( < .001) with Cognitive Fatigue belonged to genes linked to inflammation (false discovery rate adjusted = .03), cognitive disorders ( = 1.51 × 10), and synaptic transmission ( = 6.28 × 10).
Genomic analyses in this large cohort of breast cancer survivors suggest a possible genetic role for severe Cognitive Fatigue that warrants further exploration.
我们旨在利用机器学习方法,基于临床协变量和种系全基因组数据预测乳腺癌治疗后的疲劳情况。
我们获取了癌症毒性研究(NCT01993498)中2799例早期乳腺癌患者的种系全基因组数据。主要终点定义为在诊断时得分为零,且在完成初次治疗1年后,在欧洲癌症研究与治疗组织的总体疲劳生活质量问卷以及身体、情感和认知疲劳多维问卷上得分高于四分位数3。首先,我们测试了每个终点与临床变量和全基因组变异的单变量关联。然后,使用预先选定的临床变量(错误发现率<0.05)和基因组变量(<0.001),构建了一个多变量预处理随机森林回归模型,并在一个留出子集上进行验证,以预测疲劳情况。基因集富集分析确定了关键的生物学相关性(MetaCore)。所有统计检验均为双侧检验。
仅在情感和认知疲劳方面发现了具有统计学意义的临床关联,包括接受化疗、焦虑和疼痛。一些单核苷酸多态性与不同的疲劳终点有一定程度的关联(<0.001),尽管没有全基因组水平上具有统计学意义的关联(<5.00×10)。仅对于认知疲劳,基因组多变量模型的预测能力在统计学上显著优于随机模型(曲线下面积=0.59,P=0.01),并且加入临床变量后略有改善(曲线下面积=0.60,P=0.005)。发现与认知疲劳相关(<0.001)的单核苷酸多态性属于与炎症(错误发现率调整后P=0.03)、认知障碍(P=1.51×10)和突触传递(P=6.28×10)相关的基因。
在这个大型乳腺癌幸存者队列中的基因组分析表明,严重认知疲劳可能存在遗传因素,值得进一步探索。