Kang Pei-Lin, Liu Zhi-Pan
Collaborative Innovation Center of Chemistry for Energy Material, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Key Laboratory of Computational Physical Science, Department of Chemistry, Fudan University, Shanghai 200433, China.
iScience. 2020 Dec 30;24(1):102013. doi: 10.1016/j.isci.2020.102013. eCollection 2021 Jan 22.
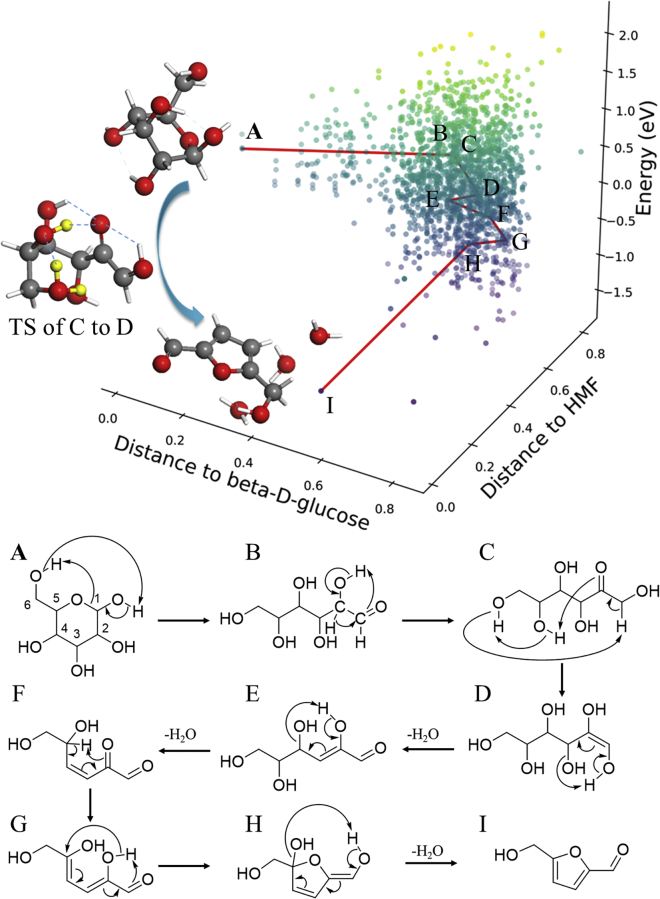
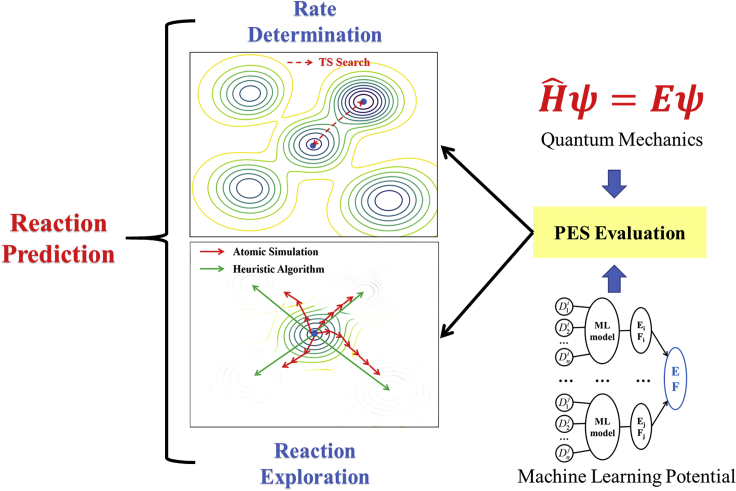
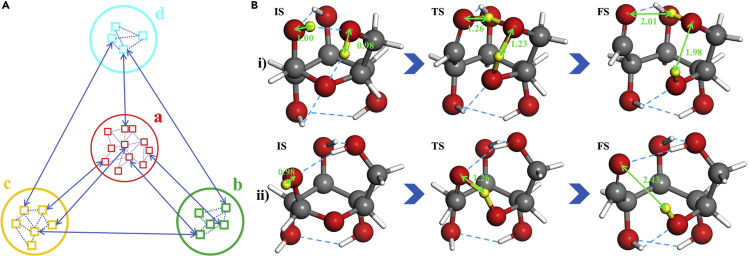
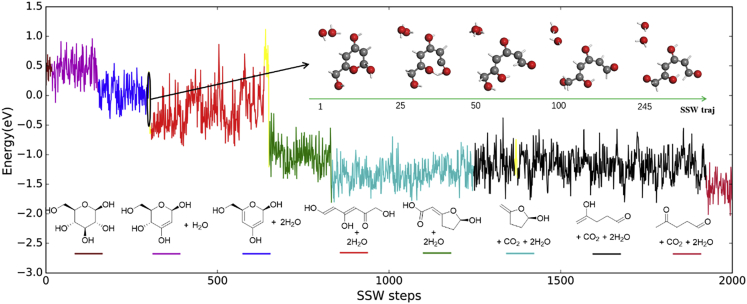
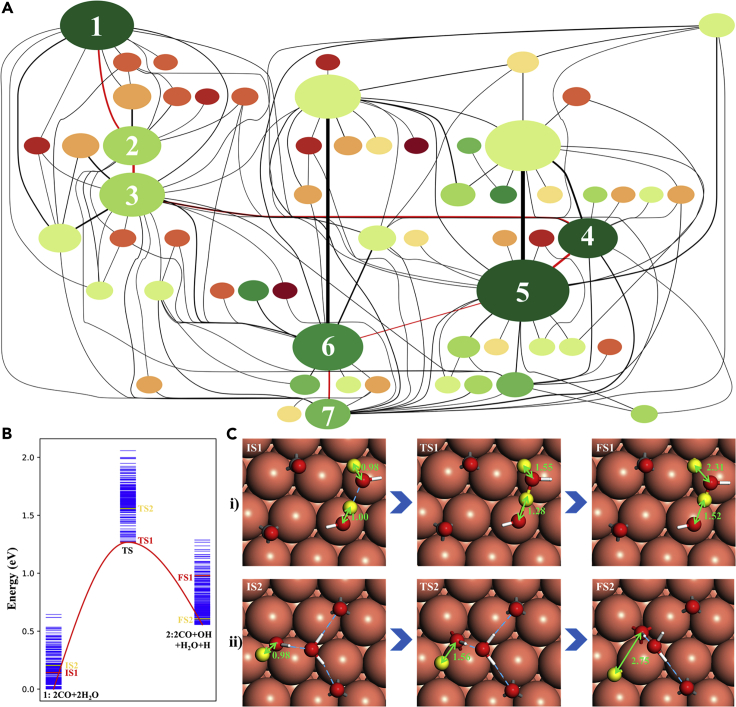
It is an ultimate goal in chemistry to predict reaction without recourse to experiment. Reaction prediction is not just the reaction rate determination of known reactions but, more broadly, the reaction exploration to identify new reaction routes. This review briefly overviews the theory on chemical reaction and the current methods for computing/estimating reaction rate and exploring reaction space. We particularly focus on the atomistic simulation methods for reaction exploration, which are benefited significantly by recently emerged machine learning potentials. We elaborate the stochastic surface walking global pathway sampling based on the global neural network (SSW-NN) potential, developed in our group since 2013, which can explore complex reactions systems unbiasedly and automatedly. Two examples, molecular reaction and heterogeneous catalytic reactions, are presented to illustrate the current status for reaction prediction using SSW-NN.
在不借助实验的情况下预测反应是化学领域的一个终极目标。反应预测不仅仅是确定已知反应的反应速率,更广泛地说,是探索反应以识别新的反应途径。本文简要概述了化学反应理论以及当前计算/估计反应速率和探索反应空间的方法。我们特别关注用于反应探索的原子模拟方法,这些方法因最近出现的机器学习势而受益匪浅。我们详细阐述了基于全局神经网络(SSW-NN)势的随机表面行走全局路径采样方法,该方法自2013年起由我们团队开发,能够无偏差且自动地探索复杂反应体系。给出了分子反应和多相催化反应两个例子来说明使用SSW-NN进行反应预测的现状。