Zhang Shuhao, Makoś Małgorzata Z, Jadrich Ryan B, Kraka Elfi, Barros Kipton, Nebgen Benjamin T, Tretiak Sergei, Isayev Olexandr, Lubbers Nicholas, Messerly Richard A, Smith Justin S
Department of Chemistry, Mellon College of Science, Carnegie Mellon University, Pittsburgh, PA, USA.
Theoretical Division, Los Alamos National Laboratory, Los Alamos, NM, USA.
Nat Chem. 2024 May;16(5):727-734. doi: 10.1038/s41557-023-01427-3. Epub 2024 Mar 7.
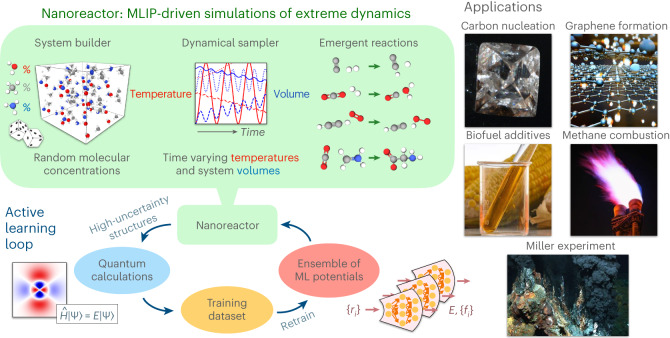
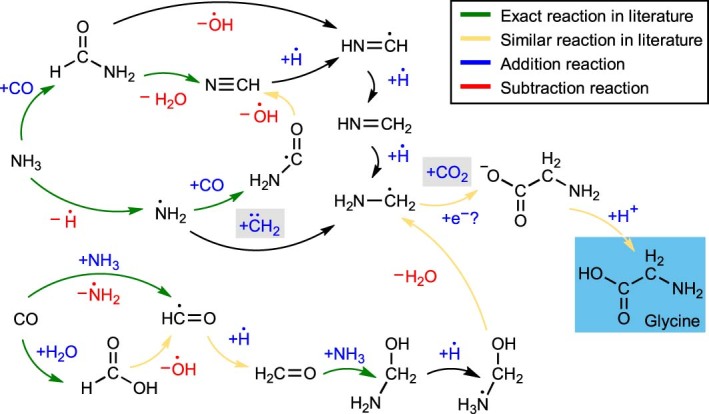
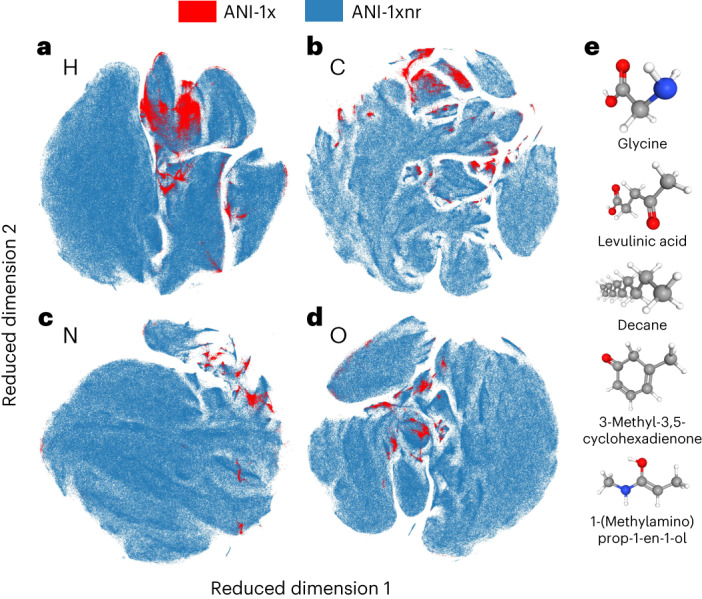
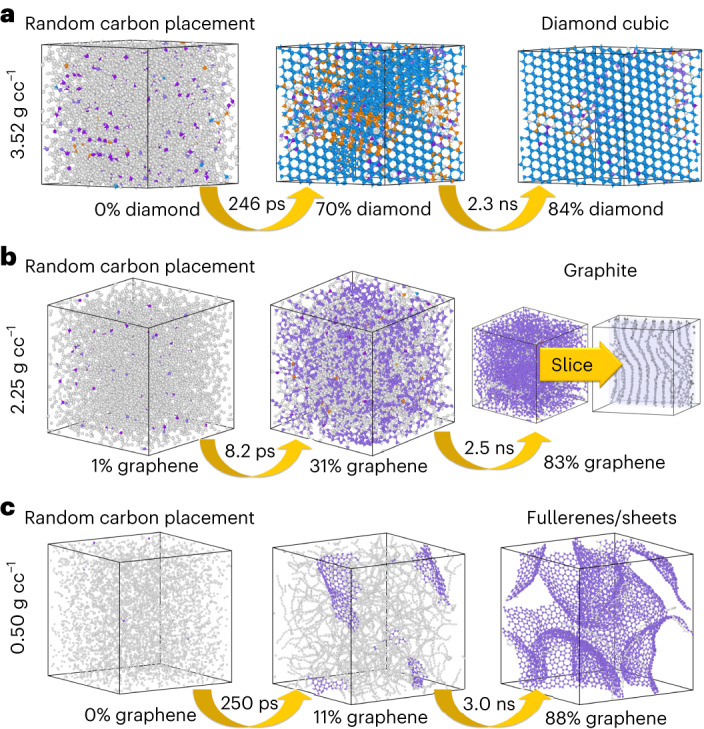
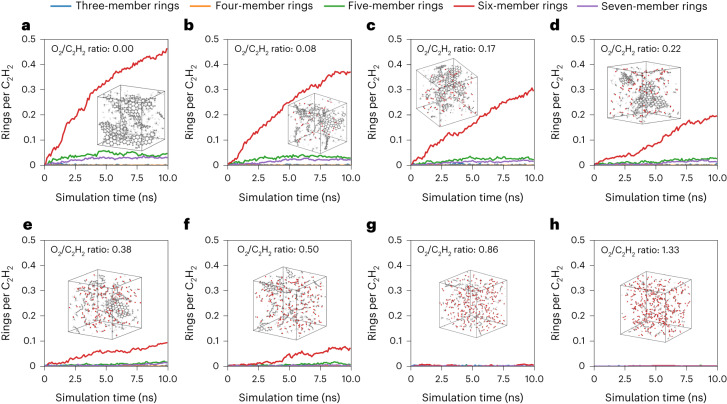
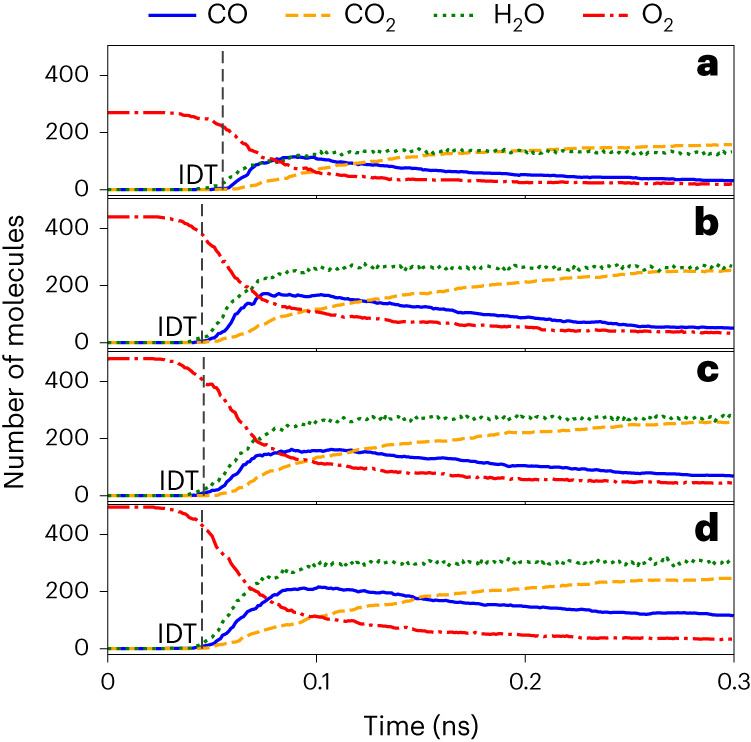
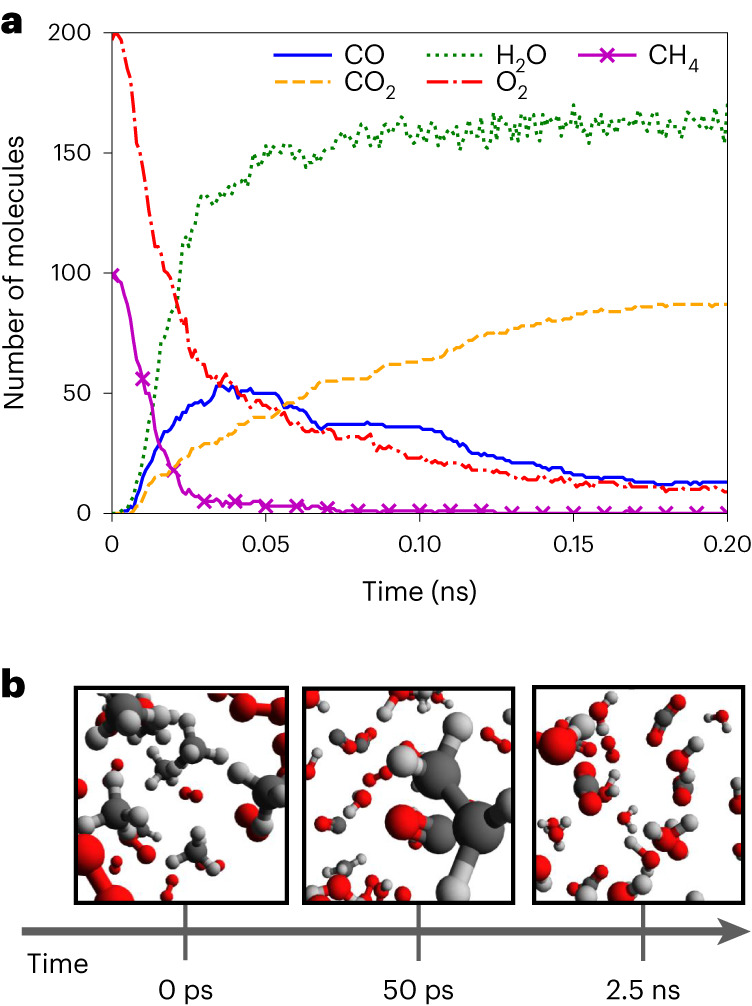
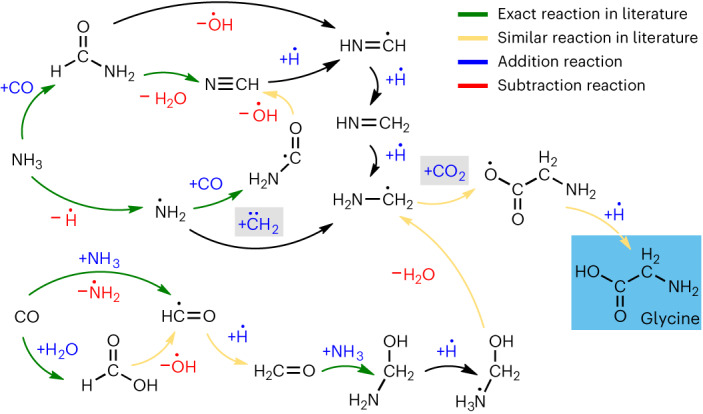
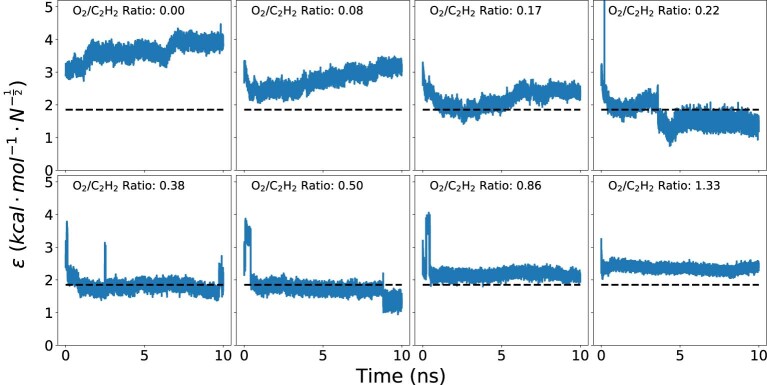
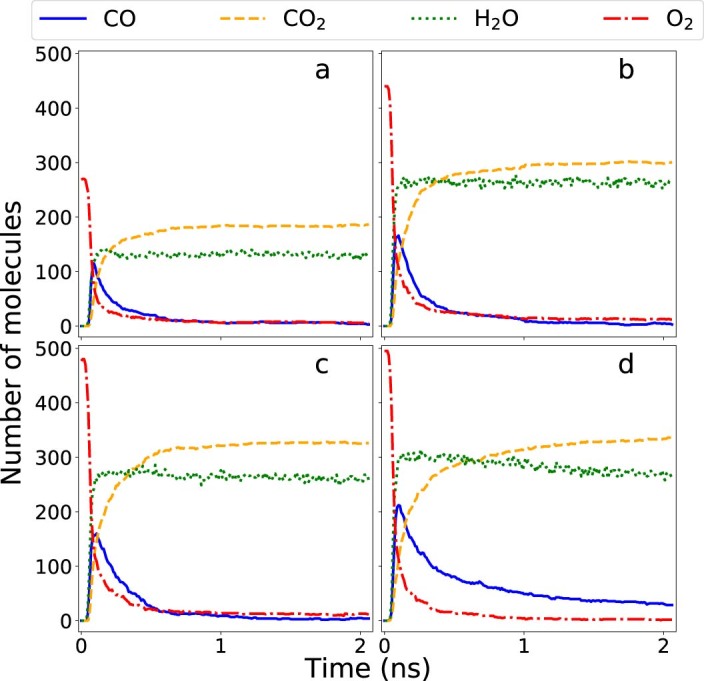
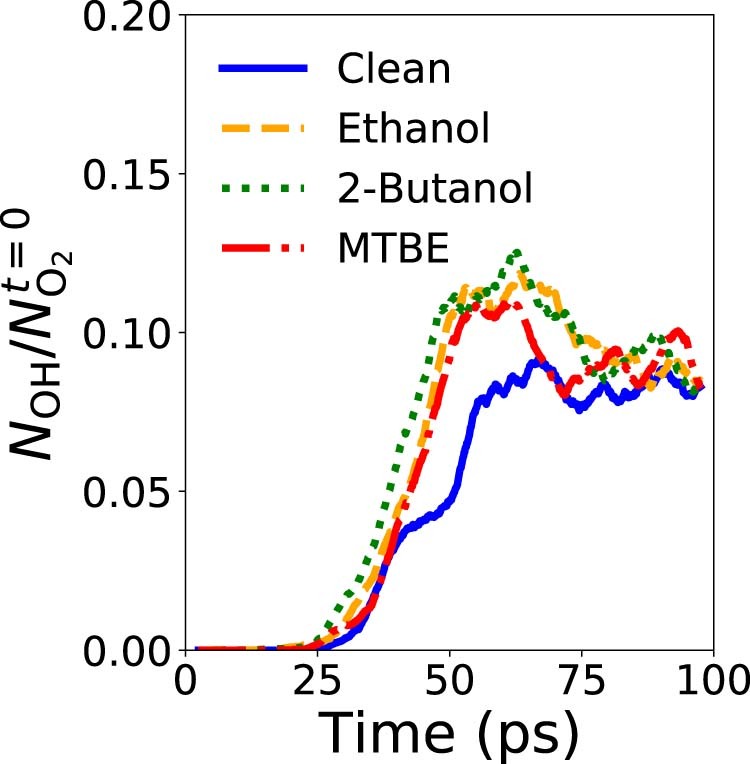
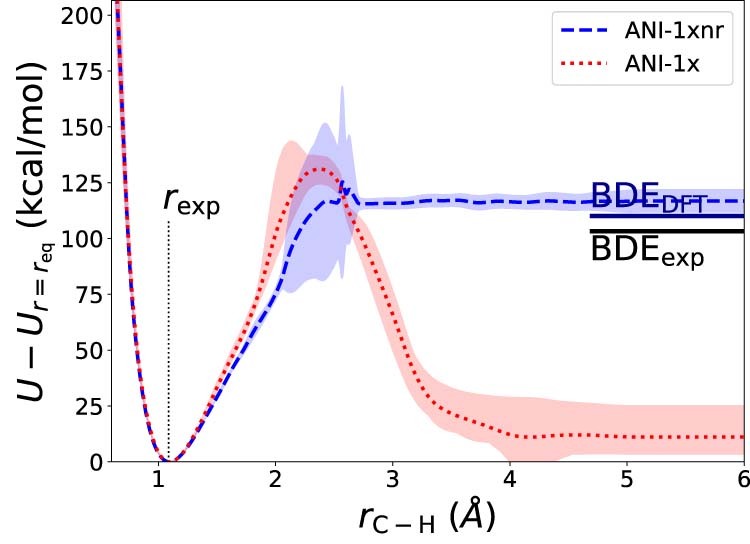
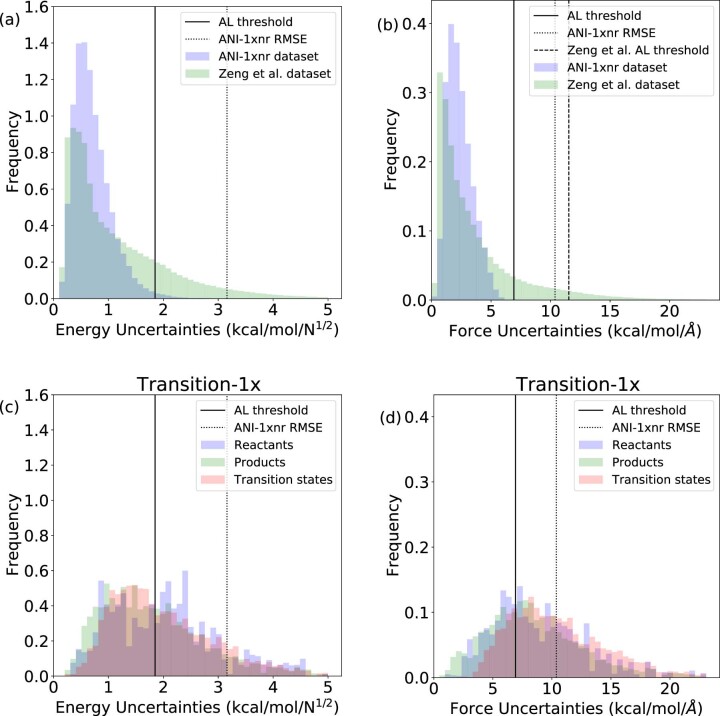
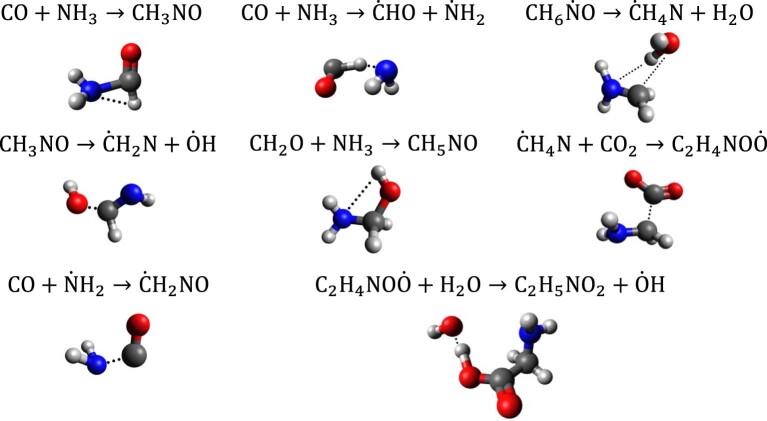
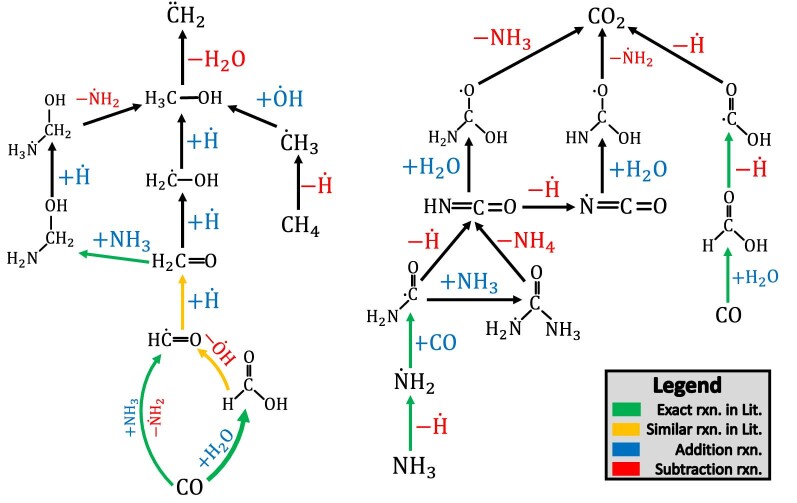
Atomistic simulation has a broad range of applications from drug design to materials discovery. Machine learning interatomic potentials (MLIPs) have become an efficient alternative to computationally expensive ab initio simulations. For this reason, chemistry and materials science would greatly benefit from a general reactive MLIP, that is, an MLIP that is applicable to a broad range of reactive chemistry without the need for refitting. Here we develop a general reactive MLIP (ANI-1xnr) through automated sampling of condensed-phase reactions. ANI-1xnr is then applied to study five distinct systems: carbon solid-phase nucleation, graphene ring formation from acetylene, biofuel additives, combustion of methane and the spontaneous formation of glycine from early earth small molecules. In all studies, ANI-1xnr closely matches experiment (when available) and/or previous studies using traditional model chemistry methods. As such, ANI-1xnr proves to be a highly general reactive MLIP for C, H, N and O elements in the condensed phase, enabling high-throughput in silico reactive chemistry experimentation.
原子模拟在从药物设计到材料发现等广泛领域都有应用。机器学习原子间势(MLIPs)已成为计算成本高昂的从头算模拟的一种有效替代方法。因此,化学和材料科学将从一种通用的反应性MLIP中大大受益,即一种无需重新拟合就能适用于广泛反应化学的MLIP。在此,我们通过凝聚相反应的自动采样开发了一种通用的反应性MLIP(ANI-1xnr)。然后将ANI-1xnr应用于研究五个不同的系统:碳的固相成核、乙炔形成石墨烯环、生物燃料添加剂、甲烷燃烧以及早期地球小分子自发形成甘氨酸。在所有研究中,ANI-1xnr与实验结果(若有)和/或之前使用传统模型化学方法的研究结果高度匹配。因此,ANI-1xnr被证明是一种用于凝聚相中碳、氢、氮和氧元素的高度通用的反应性MLIP,能够实现高通量的计算机模拟反应化学实验。