MEPHI, APHM, IRD 198, Aix Marseille Université, Institut Hospitalo-Universitaire Méditerranée Infection, 13005 Marseille, France.
Aix Marseille Université, Université de Toulon, Centre National de la Recherche Scientifique, Institut de Recherche pour le Développement, Mediterranean Institute of Oceanography UM 110, 13288 Marseille, France.
Viruses. 2021 Jan 20;13(2):148. doi: 10.3390/v13020148.
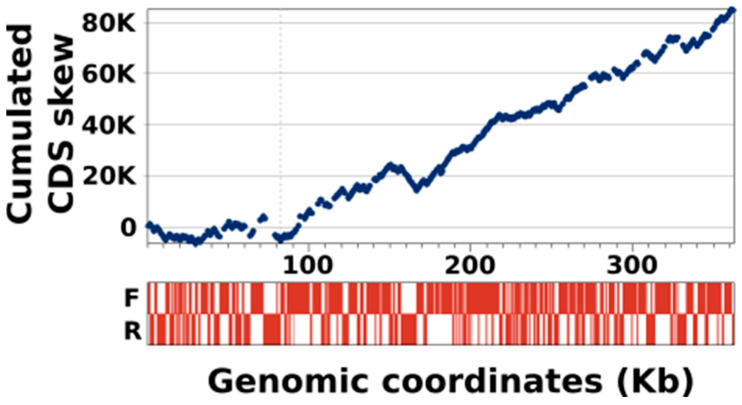
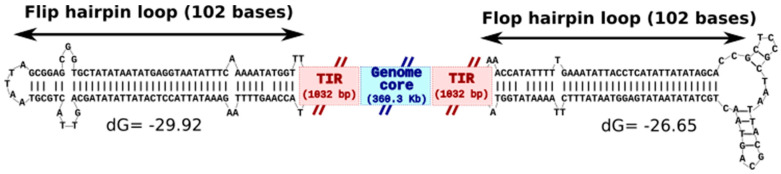
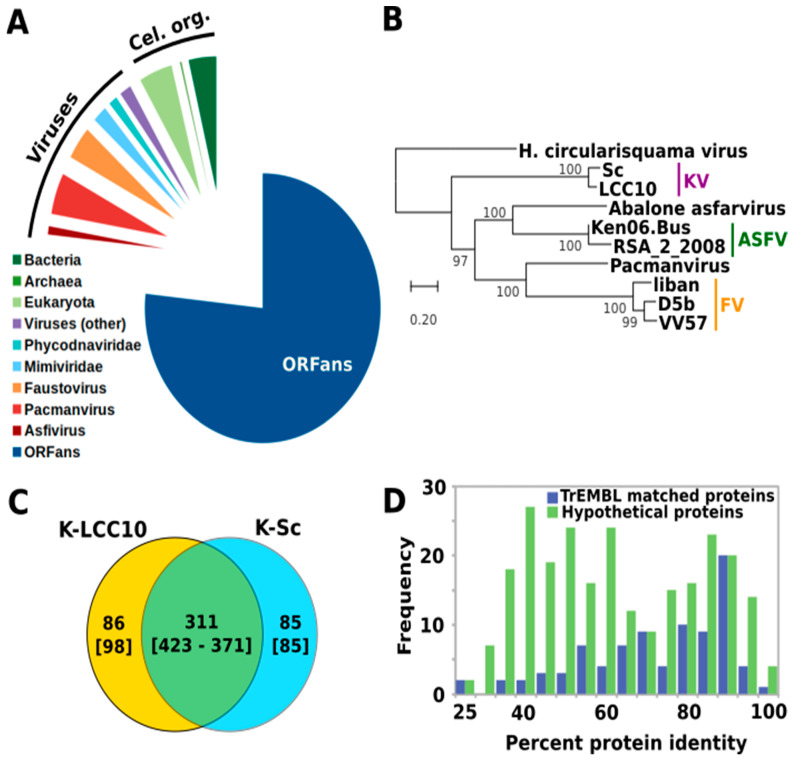
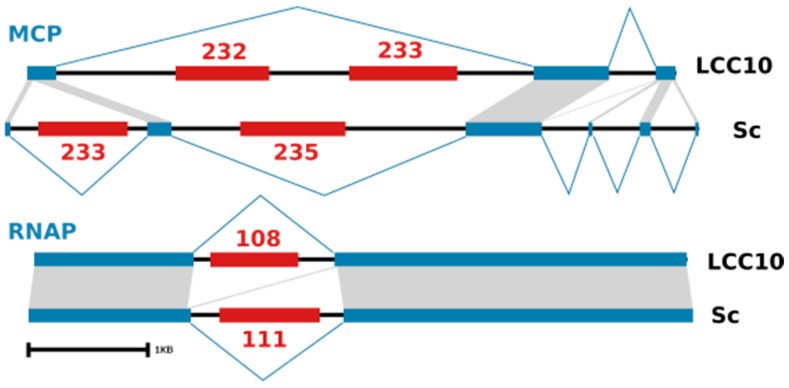
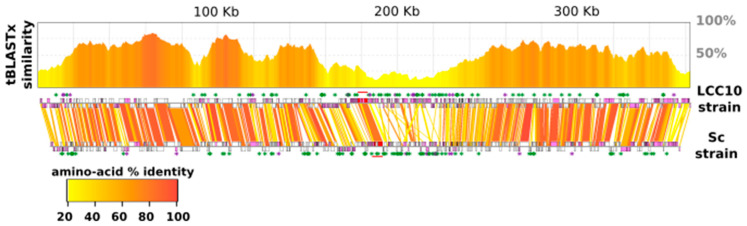
infects the amoeba and has recently been described as a distant relative of the African swine fever virus. To characterize the diversity and evolution of this novel viral genus, we report here on the isolation and genome sequencing of a second strain of , namely LCC10. Detailed analysis of the sequencing data suggested that its 362-Kb genome is linear with covalently closed hairpin termini, so that DNA forms a single continuous polynucleotide chain. Comparative genomic analysis indicated that although the two sequenced strains share extensive gene collinearity, 180 predicted genes were either gained or lost in only one genome. As already observed in another distant relative, i.e., , which infects the same host, the center and extremities of the genome exhibited a higher rate of sequence divergence and the major capsid protein gene was colonized by type-I introns. A possible role of the host in the genesis of these evolutionary traits is hypothesized. The genome exhibited a significant gene strand bias over the two-third of genome length, a feature not seen in the other members of the "extended " clade. We suggest that this gene strand bias was induced by a putative single origin of DNA replication located near the genome extremity that imparted a selective force favoring the genes positioned on the leading strand.
感染变形虫,并最近被描述为非洲猪瘟病毒的远亲。为了描述这个新的病毒属的多样性和进化,我们在这里报告了第二种的分离和基因组测序,即 LCC10。对测序数据的详细分析表明,其 362-Kb 基因组是线性的,具有共价闭合的发夹末端,因此 DNA 形成单个连续的多核苷酸链。比较基因组分析表明,尽管这两个测序的菌株共享广泛的基因共线性,但只有一个基因组中获得或丢失了 180 个预测基因。正如已经在另一个远亲中观察到的,即感染相同宿主的 ,的中心和末端表现出更高的序列分化率,主要衣壳蛋白基因被 I 型内含子殖民化。假设宿主在这些进化特征的产生中可能发挥了作用。基因组在基因组长度的三分之二以上表现出显著的基因链偏向性,这在“扩展”枝系的其他成员中没有出现。我们认为,这种基因链偏向性是由位于基因组末端附近的假定的 DNA 复制单一起源引起的,这种起源赋予了位于前导链上的基因选择性优势。